DT-061 (also known as DT061; SMAP) is a novel, potent and orally bioavailable activator of PP2A (protein phosphatase 2A) with anticancer effects. It could be applied in the therapy of KRAS-mutant and MYC-driven tumorigenesis. Treatment with DT-061, in combination with the MEK inhibitor AZD6244, resulted in suppression of both p-AKT and MYC, as well as tumor regression in two KRAS-driven lung cancer mouse models. DT-061 therapy also abrogated MYC-driven tumorigenesis.
Physicochemical Properties
| Molecular Formula | C25H23F3N2O5S |
| Molecular Weight | 520.5207 |
| Exact Mass | 520.127 |
| CAS # | 1809427-19-7 |
| Related CAS # | (1S,2S,3R)-DT-061;1809427-20-0 |
| PubChem CID | 91885558 |
| Appearance | Off-white to light yellow solid powder |
| LogP | 5.4 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 10 |
| Rotatable Bond Count | 5 |
| Heavy Atom Count | 36 |
| Complexity | 824 |
| Defined Atom Stereocenter Count | 3 |
| SMILES | S(C1C([H])=C([H])C(=C([H])C=1[H])OC(F)(F)F)(N([H])[C@]1([H])C([H])([H])C([H])([H])C([H])([H])[C@@]([H])([C@@]1([H])O[H])N1C2=C([H])C([H])=C([H])C([H])=C2OC2=C([H])C([H])=C([H])C([H])=C12)(=O)=O |
| InChi Key | WGKGADVPRVLHHZ-ZHRMCQFGSA-N |
| InChi Code | InChI=1S/C25H23F3N2O5S/c26-25(27,28)35-16-12-14-17(15-13-16)36(32,33)29-18-6-5-9-21(24(18)31)30-19-7-1-3-10-22(19)34-23-11-4-2-8-20(23)30/h1-4,7-8,10-15,18,21,24,29,31H,5-6,9H2/t18-,21+,24+/m1/s1 |
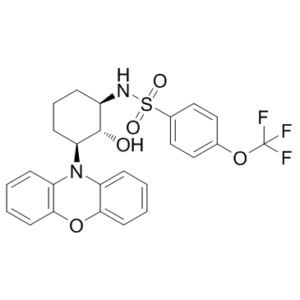
| Chemical Name | N-((1R,2R,3S)-2-hydroxy-3-(10H-phenoxazin-10-yl)cyclohexyl)-4-(trifluoromethoxy)benzenesulfonamide |
| Synonyms | DT 061; DT61; SMAP; DT-061; DT061; DT-61; DT 61 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
PP2A[1]. DT-061 directly binds to the scaffolding A subunit of protein phosphatase 2A (PP2A), specifically PPP2R1A, acting as a small-molecule activator of PP2A (SMAP) [1] |
| ln Vitro |
DT-061 showed a sensitivity profile in KRAS-mutant lung cancer cell lines overexpressing PP2A inhibitory proteins (PIPs), similar to cells with a PPP2R1A E64D mutation [1] CIP2A overexpression did not confer further resistance to DT-061 [1] In KRAS-mutant H358 and H441 cell lines, DT-061 showed dose-dependent inhibition of colony growth as a single agent [1] DT-061 in combination with the MEK inhibitor AZD6244 (at concentrations that alone did not affect colony growth) resulted in very apparent combinatorial growth inhibition in H358 and H441 cells [1] DT-061 treatment induced a dose-dependent activation of caspase-3/7 in H358 and H441 cells at a 24-hour time point, while AZD6244 alone did not [1] When both drugs were used at 10 µM, DT-061 enhanced the apoptosis response in H358 and H441 cells [1] The combination of DT-061 and MEK inhibitor potently inhibited p-AKT and MYC expression in KRAS-mutant lung cancer cell lines H358 and H441 [1] DT-061 inhibits AKT and RPS6K signaling in H358 cells [1] |
| ln Vivo |
DT-061 (5 mg/kg, oral gavage, 4 weeks) inhibits the growth of H358 or H441 xenografts as a single agent. Furthermore, there is a considerable increase in efficiency when DT-061 and AZD6244 are combined[1]. The capacity of DT-061 to inhibit H358 xenograft growth was dependent on drug binding to PPP2R1A [1] In H358 xenograft models, both DT-061 and AZD6244 showed single-agent activity in inhibiting tumor volume, but their combination was significantly more efficient than either compound alone [1] Similar results were obtained in H441 xenograft models, where the combination of DT-061 and AZD6244 was significantly more efficient than monotherapies [1] Based on RECIST criteria, most mice treated with DT-061 alone had progressive disease, while all mice treated with the combination of DT-061 and AZD6244 showed at least a partial response; one H358 xenograft showed a complete response [1] The combination of DT-061 and AZD6244 had an additive effect on reducing tumor cell proliferation (measured by PCNA-positive cells) and increasing apoptosis (measured by TUNEL-positive cells) in tumor tissue [1] DT-061 combined with AZD6244 inhibited endogenous MYC expression in drug-treated tumors in vivo [1] In H358-GFP control xenografts, DT-061 monotherapy (5 mg/kg) inhibited tumor growth over a 20-day period, with an effect comparable to the combination of AZD6244 and the AKT inhibitor MK2206 [1] In H358 xenografts overexpressing wild-type MYC, DT-061 monotherapy (5 mg/kg) inhibited tumor growth to a similar extent as the AZD6244+MK2206 combination [1] In H358 xenografts overexpressing a MYC mutant (S62D) resistant to PP2A-mediated degradation, DT-061 monotherapy (5 mg/kg) induced only partial tumor growth inhibition, whereas the AZD6244+MK2206 combination remained efficient [1] |
| Cell Assay |
For colony formation assays involving DT-061 and AZD6244, cells (H358, H441) were seeded at 1000 cells per well and allowed to grow for 3 weeks in the presence of vehicle control or increasing concentrations of the drugs. Cells were then fixed, stained with crystal violet, and quantified [1] For cell viability assays, cells were plated in 96-well plates. After incubation for 24 hours, cultures were treated with drugs for 3 days. The proportion of viable cells was determined by WST1 assay according to the manufacturer's recommendations [1] For apoptosis assays (Caspase-3/7 activity), cells were plated in 96-well plates. After incubation for 24 hours, cultures were treated with drugs. Caspase-3/7 activity was determined using a Caspase-Glo 3/7 Assay according to the manufacturer's recommendations [1] For Western blot analysis to assess signaling pathways (e.g., p-AKT, MYC), cells were treated with drugs, lysed, and proteins were analyzed using standard immunoblotting techniques [1] |
| Animal Protocol |
Animal/Disease Models: 6 - to 8weeks old male BALB/c nu/nu (nude) mice injected with H441 cells (5 × 106)[1]. Doses: 5 mg/kg. Route of Administration: po (oral gavage) for 4 weeks. Experimental Results: demonstrated activity in inhibiting tumor growth. For xenograft studies, H358 (1 × 10⁷ cells) or H441 (5 × 10⁶ cells) were injected subcutaneously into the right flank of 6- to 8-week-old male BALB/c nu/nu mice [1] When tumor volumes reached an average of 100 mm³, mice were randomized to treatment groups [1] Mice were treated by oral gavage with vehicle control, AZD6244 (25 mg/kg), DT-061 (5 mg/kg), or DT-061 in combination with AZD6244 [1] Tumor volume was assessed by caliper measurement every other day [1] Body weights were recorded weekly [1] Animals were observed for signs of toxicity (mucous diarrhea, abdominal stiffness, or weight loss) [1] Blood and tumor tissue were harvested 2 hours after the final dose [1] |
| References |
[1]. PP2A inhibition is a druggable MEK inhibitor resistance mechanism in KRAS-mutant lung cancer cells. Sci Transl Med. 2018 Jul 18;10(450). pii: eaaq1093. |
| Additional Infomation |
DT-061 is an orally bioavailable small-molecule activator of PP2A (SMAP) and a reengineered derivative of tricyclic neuroleptics [1] Mutation of the putative DT-061 target-binding site on PPP2R1A or overexpression of viral PIP SV40 small t antigen abrogates its anticancer activities [1] DT-061 is proposed to transform the MEK inhibitor response in KRAS-mutant cells from cytostatic to cytotoxic [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~125 mg/mL (~240.14 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (4.00 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: 2.08 mg/mL (4.00 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.08 mg/mL (4.00 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.9212 mL | 9.6058 mL | 19.2116 mL | |
| 5 mM | 0.3842 mL | 1.9212 mL | 3.8423 mL | |
| 10 mM | 0.1921 mL | 0.9606 mL | 1.9212 mL |