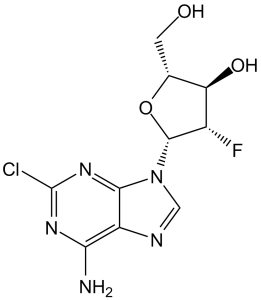
Clofarabine(formerly C1 F-Ara-A; C1 F-Ara-A; CAFdA; trade names: Clofarex; Clolar) is an antimetabolite anticancer chemotherapeutic drug and a purine nucleoside approved for the treatment of relapsed or refractory acute lymphoblastic leukemia. It functions as both a substrate of Deoxycytidine Kinase (dCK) and an inhibitor of DNA synthesis. DNA polymerase-α and -ε are competed with by clofarabine triphosphate, which is produced when clofarabine is phosphorylated. DNA elongation and repair are hampered when clofarabine-monophosphate is incorporated into internal and terminal DNA sites at the same time. With an IC50 value of 65 nM, clofarabine triphosphate inhibits ribonucleotide reductase, reducing dCTP and dATP in the process. Through the nucleoside transporters hENT1, hENT2, and hCNT2, clofarabine is effectively incorporated into cells.
Physicochemical Properties
| Molecular Formula | C10H11CLFN5O3 | |
| Molecular Weight | 303.68 | |
| Exact Mass | 303.053 | |
| Elemental Analysis | C, 39.55; H, 3.65; Cl, 11.67; F, 6.26; N, 23.06; O, 15.81 | |
| CAS # | 123318-82-1 | |
| Related CAS # |
|
|
| PubChem CID | 119182 | |
| Appearance | White to off-white solid powder | |
| Density | 2.1±0.1 g/cm3 | |
| Boiling Point | 599.5±60.0 °C at 760 mmHg | |
| Melting Point | 228-2310C | |
| Flash Point | 316.4±32.9 °C | |
| Vapour Pressure | 0.0±1.8 mmHg at 25°C | |
| Index of Refraction | 1.844 | |
| LogP | 0.24 | |
| Hydrogen Bond Donor Count | 3 | |
| Hydrogen Bond Acceptor Count | 8 | |
| Rotatable Bond Count | 2 | |
| Heavy Atom Count | 20 | |
| Complexity | 370 | |
| Defined Atom Stereocenter Count | 4 | |
| SMILES | ClC1=NC(=C2C(=N1)N(C([H])=N2)[C@@]1([H])[C@]([H])([C@@]([H])([C@@]([H])(C([H])([H])O[H])O1)O[H])F)N([H])[H] |
|
| InChi Key | WDDPHFBMKLOVOX-AYQXTPAHSA-N | |
| InChi Code | InChI=1S/C10H11ClFN5O3/c11-10-15-7(13)5-8(16-10)17(2-14-5)9-4(12)6(19)3(1-18)20-9/h2-4,6,9,18-19H,1H2,(H2,13,15,16)/t3-,4+,6-,9-/m1/s1 | |
| Chemical Name | (2R,3R,4S,5R)-5-(6-amino-2-chloropurin-9-yl)-4-fluoro-2-(hydroxymethyl)oxolan-3-ol | |
| Synonyms | C1 F-Ara-A; C1-F-Ara-A; Clofarabine; C1 F-Ara-A; trade names: Clofarex; Clolar. Abbreviation: CAFdA; 123318-82-1; Evoltra; Clofarex; CAFdA; Cl-F-Ara-A; C1-F-Ara-A; | |
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | Ribonucleotide reductase ( IC50 = 65 nM ) | |
| ln Vitro |
|
|
| ln Vivo |
|
|
| Cell Assay |
Cell Line: NB4 cells Concentration: 0.01-0.1 µM Incubation Time: 48 hours Result: Inhibited proliferation of NB4 cells in a concentration-depended manner. K562 cells were incubated with clofarabine and ara-C either sequentially or simultaneously to evaluate the combination effect on their phosphorylated metabolites. Clonogenic assays were used to determine the cytotoxicity of each agent alone and in combination. Deoxynucleotide analysis was performed to assess the effect of clofarabine on dNTPs.[3] |
|
| Animal Protocol |
Kunming mice (18-22 g, with equal numbers of male and female mice) 600, 480, 384, 307, 246 mg/kg Injected intraperitoneally at 8:00 am, 12:00 noon, 8:00 pm and 12:00 midnight; 7 days continuous administration To evaluate the time- and dose-dependent toxicity of clofarabine in mice and to further define the chronotherapy strategy of it in leukemia, we compared the mortality rates, LD50s, biochemical parameters, histological changes and organ indexes of mice treated with clofarabine at various doses and time points. Plasma clofarabine levels and pharmacokinetic parameters were monitored continuously for up to 8 hours after the single intravenous administration of 20 mg/kg at 12:00 noon and 12:00 midnight by high performance liquid chromatography (HPLC)-UV method. Clofarabine toxicity in all groups fluctuated in accordance with circadian rhythms in vivo. The toxicity of clofarabine in mice in the rest phase was more severe than the active one, indicated by more severe liver damage, immunodepression, higher mortality rate, and lower LD50. No significant pharmacokinetic parameter changes were observed between the night and daytime treatment groups. These findings suggest the dosing-time dependent toxicity of clofarabine synchronizes with the circadian rhythm of mice, which might provide new therapeutic strategies in further clinical application.[4] |
|
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion Based on 24-hour urine collections in the pediatric studies, 49 - 60% of the dose is excreted in the urine unchanged. 172 L/m2 28.8 L/h/m2 [Pediatric patients (2 - 19 years old) with relapsed or refractory acute lymphoblastic leukemia (ALL) or acute myelogenous leukemia (AML) receiving 52 mg/m2 dose] Metabolism / Metabolites Clofarabine is sequentially metabolized intracellularly to the 5’-monophosphate metabolite by deoxycytidine kinase and mono- and di-phosphokinases to the active 5’-triphosphate metabolite. Clofarabine has high affinity for the activating phosphorylating enzyme, deoxycytidine kinase, equal to or greater than that of the natural substrate, deoxycytidine. Biological Half-Life The terminal half-life is estimated to be 5.2 hours. |
|
| Toxicity/Toxicokinetics |
Hepatotoxicity In clinical trials, serum enzymes elevations occurred in up to 75% of patients treated with clofarabine as monotherapy for refractory or relapsed acute leukemia. These elevations usually arose within 5 to 10 days of starting therapy and were generally transient and asymptomatic. The elevations rarely required dose adjustment or delay in therapy. Cases of clinically apparent liver injury due to clofarabine have been reported to occur, but few details were available and most patients were receiving other cancer chemotherapeutic agents. A single case report of toxic epidermal necrosis and fulminant hepatic failure in a child with ALL receiving clofarabine has been published. In high doses, clofarabine has been associated with very high rates of serum enzyme elevations and hyperbilirubinemia that are dose limiting. Instances of capillary leak syndrome and possibly sinusoidal obstruction syndrome have also been reported. Likelihood score: D (possible rare cause of clinically apparent liver injury). Protein Binding 47% bound to plasma proteins, predominantly to albumin. |
|
| References |
[1]. Nat Rev Drug Discov . 2006 Oct;5(10):855-63. [2]. Clin Cancer Res . 2001 Nov;7(11):3580-9. [3]. Cancer Chemother Pharmacol . 2005 Apr;55(4):361-368. [4]. Kaohsiung J Med Sci. 2016 May;32(5):227-34. |
|
| Additional Infomation |
Pharmacodynamics Clofarabine is a purine nucleoside antimetabolite that differs from other puring nucleoside analogs by the presence of a chlorine in the purine ring and a flourine in the ribose moiety. Clofarabine seems to interfere with the growth of cancer cells, which are eventually destroyed. Since the growth of normal body cells may also be affected by clofarabine, other effects also occur. Clofarabine prevents cells from making DNA and RNA by interfering with the synthesis of nucleic acids, thus stopping the growth of cancer cells. |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (6.85 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (6.85 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.08 mg/mL (6.85 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.2929 mL | 16.4647 mL | 32.9294 mL | |
| 5 mM | 0.6586 mL | 3.2929 mL | 6.5859 mL | |
| 10 mM | 0.3293 mL | 1.6465 mL | 3.2929 mL |