Physicochemical Properties
| Molecular Formula | C23H31CLFN3O5 |
| Molecular Weight | 483.960748910904 |
| Exact Mass | 483.193 |
| Elemental Analysis | C, 57.08; H, 6.46; Cl, 7.32; F, 3.93; N, 8.68; O, 16.53 |
| CAS # | 260779-88-2 |
| Related CAS # | Cisapride;81098-60-4; 60779-88-2 (monohydrate); 189888-25-3 (tartrate) |
| PubChem CID | 6917697 |
| Appearance | Typically exists as solid at room temperature |
| Hydrogen Bond Donor Count | 3 |
| Hydrogen Bond Acceptor Count | 8 |
| Rotatable Bond Count | 9 |
| Heavy Atom Count | 33 |
| Complexity | 581 |
| Defined Atom Stereocenter Count | 2 |
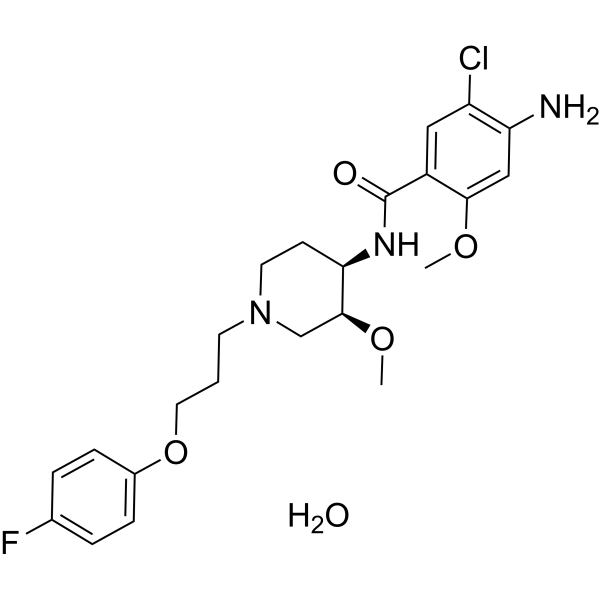
| SMILES | CO[C@H]1CN(CC[C@H]1NC(=O)C2=CC(=C(C=C2OC)N)Cl)CCCOC3=CC=C(C=C3)F.O |
| InChi Key | QBYYXIDJOFZORM-LBPAWUGGSA-N |
| InChi Code | InChI=1S/C23H29ClFN3O4.H2O/c1-30-21-13-19(26)18(24)12-17(21)23(29)27-20-8-10-28(14-22(20)31-2)9-3-11-32-16-6-4-15(25)5-7-16;/h4-7,12-13,20,22H,3,8-11,14,26H2,1-2H3,(H,27,29);1H2/t20-,22+;/m1./s1 |
| Chemical Name | 4-amino-5-chloro-N-[(3S,4R)-1-[3-(4-fluorophenoxy)propyl]-3-methoxypiperidin-4-yl]-2-methoxybenzamide;hydrate |
| Synonyms | Cisapride monohydrate; Alimix; Cisapride hydrate; MFCD03305346; Acenalin; Propulsid; 4-AMINO-5-CHLORO-N-((3R,4S)-1-(3-(4-FLUOROPHENOXY)PROPYL)-3-METHOXYPIPERIDIN-4-YL)-2-METHOXYBENZAMIDE HYDRATE; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | 5-HT4 receptor ( EC50 = 140 nM ); hERG channel ( IC50 = 9.4 nM ) |
| ln Vitro | Cisapride is an agonist of the 5-HT4 receptor and an hERG blocker[1]. It is frequently used to treat disorders of gastrointestinal motility, including gastroparesis, slow-transit constipation, and chronic intestinal pseudo-obstruction, or gastroesophageal reflux disease (GERD). Through its ability to stimulate intestinal acetylcholine release from muscarinic receptors, cisapride works to promote motility of the gastrointestinal tract. Cisapride blocks the current in ventricular myocytes' L-type Ca2+ channels and the Kv 4.3 channels that are consistently expressed in Chinese hamster ovary (CEO) cells. In a concentration-dependent manner, cisapride inhibited Kv channel current without affecting its ability to act as a selective agonist of the serotonin 5-HT4 receptor[2]. |
| ln Vivo | Cisapride (0.1-1 mg/kg; injection, once) stimulates antral and aqueous motility in conscious dogs [3]. Endpoints of cisapride (2 mg/kg, (ip); 4 mg/kg, (oral); once) versus trinitrobenzene sulfonic acid treatment endpoints in terms of macroscopic features, histopathological features, cytokine profiles, and body weight changes There is no significant difference [4]. Animal model: Rat colitis induced by trinitrobenzene sulfonic acid (TNBS) in male Wistar rats [4] Dosage: 2 mg/kg (intraperitoneal injection); 4 mg/kg (orally) Administration method: 2 mg/kg, intraperitoneal injection; 4 mg/kg, oral administration; Results: Colitis rats developed severe and intense transmural inflammation and diffuse necrosis, inflammatory granuloma and submucosal neutrophil infiltration. Cause weight loss. There is a pressing need for research that will lead to the reveal of targets designed to analyse the possible pathways for the treatment of IBD. Because of the probable involvement of serotonin in inflammatory conditions of intestine and the important role of 5HT(4) receptors in GI function, the investigation of the role of 5HT(4) receptors in the pathogenesis of IBD will be interesting. The aim of this study was to investigate the effects of cisapride, a 5HT(4) receptor agonist, in trinitrobenzenesulfonic-acid-(TNBS) induced rat colitis. Two hours subsequent to induction of colitis using TNBS in rats, cisapride (2 mg/kg, intraperitoneally (i.p); 4 mg/kg, orally (p.o)) and dexamethasone (1 mg/kg, i.p; 2 mg/kg, p.o) were administrated for 6 days. Animals were thereafter euthanized; macroscopic, histological, and biochemical assessments and ELISA test were carried out on distal colon samples. Our data showed that dexamethasone treatment (i.p, p.o) significantly decreased macroscopic and microscopic damage and also biochemical markers, but there were no significant differences in aforementioned parameters between cisapride (i.p or p.o) and TNBS-treated rats. It can be deduced that because the severity of colitis produced by TNBS is massive (through various pathways), cisapride could not bring about more colitis damages through 5HT(4) receptors. Based on the present study further researches are required for investigating the exact roles of 5HT(4) receptors in the pathogenesis of ulcerative colitis.[4] |
| Enzyme Assay | Mosapride and cisapride are gastroprokinetic agents with 5-hydroxytryptamine4 receptor agonist activity and have been widely used in the treatment of a variety of gastrointestinal disorders. The effects of mosapride and cisapride on cloned Kv4.3 channels stably expressed in Chinese hamster ovary cells were investigated using the whole-cell patch-clamp technique. Mosapride and cisapride inhibited Kv4.3 in a concentration-dependent manner with IC50 values of 15.2 and 9.8 μM, respectively. Mosapride accelerated the rate of inactivation and activation of Kv4.3 in a concentration-dependent manner and thereby decreased the time to peak. The rate constants of association (k +1) and dissociation (k -1) for mosapride were 9.9 μM(-1) s(-1) and 151.3 s(-1), respectively. The K D (k -1/k +1) was 16.2 μM, similar to the IC50 value calculated from the concentration-response curve. Voltage-dependent inhibition by mosapride was observed in the voltage range for channel opening but was not observed over a voltage range in which all Kv4.3 channels were open. Both the steady-state activation and inactivation curves of Kv4.3 were shifted in the hyperpolarizing direction in the presence of mosapride. Mosapride also caused a substantial acceleration in closed-state inactivation of Kv4.3. Mosapride produced use-dependent inhibition, which was consistent with a slow recovery from inactivation of Kv4.3. M1 and norcisapride, the major metabolites of mosapride and cisapride, respectively, had little or no effect on Kv4.3. These results indicate that mosapride inhibits Kv4.3 by both preferential binding to the open state of the channels during depolarization and acceleration of the closed-state inactivation at subthreshold potentials[2]. |
| Cell Assay | The blocking effect of three 5-HT(4) agonists, cisapride, mosapride, and the newly discovered CJ-033466 on the human ether-a-go-go-related gene (hERG) channel was studied using a whole cell patch-clamp technique in HEK293 cells. Cisapride was found to be the most potent of the hERG blockers. CJ-033466 had the widest safety margin between its hERG blocking activity and 5-HT(4) agonism among the tested compounds. This suggests a lower clinical risk of cardiac arrhythmia in CJ-033466 compared with the other 2 agonists. Therefore, CJ-033466 has the potential to be a drug with higher therapeutic efficacy and less cardiac risk than both cisapride and mosapride[1]. |
| Animal Protocol | Male Wistar rats with trinitrobenzenesulfonic-acid-(TNBS) induced rat colitis 2 mg/kg (i.p.); 4 mg/kg, (oral administration) 2 mg/kg, intraperitoneal injection ; 4 mg/kg, oral administration; once |
| ADME/Pharmacokinetics |
Absorption Cisapride is rapidly absorbed after oral administration, with an absolute bioavailability of 35-40%. The placental transfer of cisapride, a new prokinetic agent, was studied in a sheep model. The pharmacokinetics of cisapride were studied in the lamb, the pregnant ewe, and the fetus by obtaining blood samples from chronically implanted arterial catheters. Comparable pharmacokinetic parameters were found in the lamb and the adult sheep: half-life, 1.39-1.83 hr; total plasma clearance, 1998-2160 ml/kg/hr; AUC, 92.6-100.1 ng.hr/ml. Cisapride plasma concentrations after continuous infusion were predicted correctly based on the parameters obtained after IV bolus. There was a materno-fetal transfer of cisapride following a single IV bolus administered to the mother. Cisapride crossed the placenta within 5 min and equilibrated with maternal plasma within 20 to 30 min after dosing. The average fetal-to-maternal plasma concentration ratio was 0.71. The amniotic fluid also contained measurable amounts of cisapride. The protein binding of cisapride in maternal and fetal plasma is 89.0% and 88.4%, respectively; the free fraction is 4 times larger than in humans. Cisapride crosses the ovine placental barrier. The sheep placenta is less permeable than the human placenta, but the higher free fraction of cisapride facilitates placental transfer. PMID:1673393 Metabolism / Metabolites Hepatic. Extensively metabolized via cytochrome P450 3A4 enzyme. IPA COPYRIGHT: ASHP The metabolism of cisapride in vitro using Liver fractions of dogs, rabbits, and rats and the metabolites identified by high performance LC and by MS are described. Main bi otransformat i on routes were oxi dat i ve N-dealkylat i on at the pi peri di ne ni trogen and aron at i c hydroxylat i on at the fluorophenyl or at the benzami de moi ety. ENG ~21 nq~_~n_~. Biological Half-Life: 6-12 hours |
| Toxicity/Toxicokinetics |
Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Cisapride was removed from the market in the United States by the U.S. Food and Drug Administration because of cardiac toxicity. Because of the low levels of cisapride in breastmilk, its use is acceptable in nursing mothers if it is required. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. |
| References |
[1]. Wiseman, L.R., Faulds, D. Cisapride. Drugs 47, 116–152 (1994). [2]. The 5-HT(4) agonists cisapride, mosapride, and CJ-033466, a Novel potent compound, exhibit different human ether-a-go-go-related gene (hERG)-blocking activities. J Pharmacol Sci. 2007 Oct;105(2):207-10. |
| Additional Infomation |
A substituted benzamide used for its prokinetic properties. It is used in the management of gastroesophageal reflux disease, functional dyspepsia, and other disorders associated with impaired gastrointestinal motility. (Martindale The Extra Pharmacopoeia, 31st ed) See also: Cisapride (has active moiety). |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0663 mL | 10.3314 mL | 20.6629 mL | |
| 5 mM | 0.4133 mL | 2.0663 mL | 4.1326 mL | |
| 10 mM | 0.2066 mL | 1.0331 mL | 2.0663 mL |