Physicochemical Properties
| Molecular Formula | C8H16NO5P |
| Molecular Weight | 237.19 |
| Exact Mass | 237.077 |
| Elemental Analysis | C, 40.51; H, 6.80; N, 5.91; O, 33.73; P, 13.06 |
| CAS # | 146388-56-9 |
| PubChem CID | 6438792 |
| Appearance | Typically exists as solid at room temperature |
| Density | 1.312g/cm3 |
| Boiling Point | 434.1ºC at 760 mmHg |
| Flash Point | 216.3ºC |
| Vapour Pressure | 9.46E-09mmHg at 25°C |
| Index of Refraction | 1.512 |
| LogP | 0.701 |
| Hydrogen Bond Donor Count | 3 |
| Hydrogen Bond Acceptor Count | 6 |
| Rotatable Bond Count | 6 |
| Heavy Atom Count | 15 |
| Complexity | 295 |
| Defined Atom Stereocenter Count | 1 |
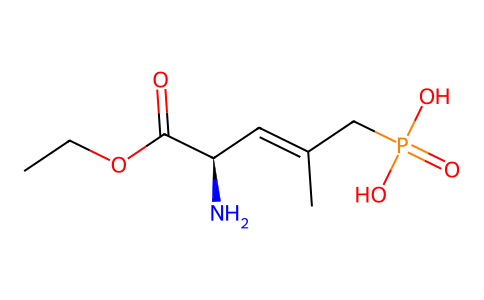
| SMILES | CCOC([C@@H](/C=C(/CP(=O)(O)O)\C)N)=O |
| InChi Key | OKDOWCKDTWNRCB-PTYLAXBQSA-N |
| InChi Code | InChI=1S/C8H16NO5P/c1-3-14-8(10)7(9)4-6(2)5-15(11,12)13/h4,7H,3,5,9H2,1-2H3,(H2,11,12,13)/b6-4+/t7-/m1/s1 |
| Chemical Name | [(E,4R)-4-amino-5-ethoxy-2-methyl-5-oxopent-2-enyl]phosphonic acid |
| Synonyms | Cgp-43487; Cgp43487; Cgp 43487; 146388-56-9; Cgp-43,487; 3-Pentenoic acid, 2-amino-4-methyl-5-phosphono-, 1-ethyl ester, (R-(E))-; [(E,4R)-4-amino-5-ethoxy-2-methyl-5-oxopent-2-enyl]phosphonic acid; (R,E)-(4-amino-5-ethoxy-2-methyl-5-oxopent-2-en-1-yl)phosphonic acid; CGP-39,551; Cgp 43487 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | NMDA/N-methyl-D-aspartate receptor |
| ln Vivo |
Nicardipine, nifedipine and flunarizine showed anticonvulsive activity (reflected by significant elevations of the seizure threshold for tonic hindlimb extension) in doses of 20, 20 and 15 mg/kg, respectively. In combination studies, CGP 40116 [D-(E)-2-amino-4-methyl-5-phosphono-3-pentenoic acid] or its methyl ester derivative (CGP 43487) was administered in a constant dose of 0.25 and 3.5 mg/kg, respectively. At these doses both competitive NMDA receptor antagonists were able to elevate significantly the convulsive threshold. Nicardipine, nifedipine, and flunarizine were administered at maximal doses (or lower) not affecting the convulsive threshold (15, 15 and 10 mg/kg, respectively). The protective activity of CGP 40116 and CGP 43487 was dose dependently potentiated by all three Ca2+ channel inhibitors. The combined treatment caused motor impairments (evaluated in the chimney test) and long-term memory deficits (measured in the passive avoidance task) similar to these produced by CGP 40116 or CGP 43487 alone. Our results indicate that nicardipine, nifedipine and flunarizine significantly potentiate the protective activity, but not the adverse effects, of CGP 40116 and CGP 43487 in mice.[1] The administration to rats of different doses of the non competitive NMDA receptor blocker MK-801 (0.03-1 mg/kg IP) induced stimulation or reduction of locomotor activity, depending on the dose, whereas the competitive NMDA antagonists CGP 43487 (0.188-6 mg/kg IP) and APV (2.5-20 micrograms/rat ICV) inhibited locomotion at the highest doses. Unlike MK-801 and APV treatment, the administration of CGP 43487 did not induce impairment of rota-rod test performance. Both competitive and non-competitive NMDA antagonists, at doses devoid of any behavioral effect per se, potentiated the responses elicited by apomorphine (0.25 mg/kg SC). In particular, the occurrence of episodes of licking was weakly affected by MK-801 administration, but significantly increased by CGP 43487 and APV treatment; the presence of gnawing was augmented by all the pretreatments; sniffing, locomotion, grooming and rearing occurrence were not affected by the administration of NMDA antagonists. The results suggest that the competitive antagonists which facilitated dopaminergic function without causing motor impairment could be useful supplements in the treatment of Parkinson's disease [2]. |
| Animal Protocol |
Locomotor activity [2] Different groups of naive rats (n = 8 12 per group) were treated with various doses of MK-801 (0.03, 0.0625, 0.125, 0.25 and 1 mg/kg IP), CGP 43487 (0.188, 0,375, 0.75, 3 and 6 mg/kg IP), or APV (2,5, 5, 10 and 20 ~tg/rat, ICV) and 30 min, 180 or 5 min later, respectively, they were placed in actometric cages (Dall'Olio et al. 1988) where their motility was recorded for 60 min. Stereotyped behavior [2] Stereotyped behavior induced by subcutaneous administration of apomorphine (0.25 mg/kg) was observed in different groups of rats (n=8-16 per group) pretreated with MK-801 (0.03 and 0.0625mg/kg IP, 30 rain before), CGP 43487 (0.375 and 0.75 mg/kg IP, 3 h before) or APV (2.5 and 5 pg/rat, ICV, 5 min before). The animals were placed in actometric cages and habituated to the experimental environment for 2 h before the dopaminomimetic injection. Starting 10 min after apomorphine administration, animals were assessed using a behavioral checklist technique (Fray et al. 1980; Molloy and Waddington 1984, 1987). Each rat was observed individually for 5-s periods at l-rain intervals over 5 consecutive minutes to determine the presence of the following individual behaviors (occurring alone or in combination): sniffing (Sn); licking (Lk); gnawing (Gn); grooming (Gr, of any form); stillness (St; motionless, with no behavior evident); locomotion (L); rearing (R). After assessment using the behavioral checklist, the animals were observed for the stereotypies (STR) using a conventional 0-6 point rating scale: 0 = asleep or inactive; 1 = episodes of normal activities; 2 = discontinuous activity with bursts of sniffing or rearing; 3 = continuous stereotyped activity such as sniffing or rearing along a fixed path; 4 = stereotyped sniffing or rearing fixated in one location; 5 = stereotyped behavior with bursts of licking or gnawing; 6 = continuous licking or gnawing. This cycle of observations was repeated three times at 10-rain intervals by observers unaware of the treatments. Rats were used only once (from 0900 hours to 1500 hours). Rota-rod test [2] A rota-rod treadmill for rats was used. Rats were trained to the apparatus on four occasions at 30 min intervals. Only the animals that were able to maintain equilibrium on the rotating rod (speed 16 rpm) for at least 10 s in all the training trials were used for assaying the drug effects. Twenty-four hours after training, rats (n = 12 per group) were observed for their rota-rod performance before receiving MK-801 (0.5 and 1 mg/kg IP), CGP 43487 (3 mg/kg IP) or APV (10 pg/rat, ICV); after 30, 180 or 5 rain, respectively, the time spent by the animals on the rota-rod was evaluated again. CGP 43487 was dissolved in saline and intraperitoneally injected. Chimney test [2] The chimney test of Boissier et al. (1960) was used to evaluate the influence of CGP 40116 or CGP 43487 alone or in combination with the Ca 2+ channel inhibitors on motor performance. Motor impairment in this test was indicated by the inability of the animals to climb backwards up the tube (3 cm inner diameter, 25 cm length) within 60 s. The animals were pretrained 24 h before treatment and those unable to perform the test were rejected from the experimental groups. On the following day, mice were treated with the compounds either alone or in combination. Results were calculated as a percentage of animals failing to perform the test. Passive avoidance acquisition and retention testing [2] According to Venault et al. (1986), the step-through passive avoidance task may be used as a measure of long-term memory. We used this test to compare the influence of CGP 40116, CGP 43487, nicardipine, nifedipine, flunarizine alone or in combination (an NMDA receptor antagonist+ a Ca 2- channel inhibitor) on passive avoidance acquisition in mice. Procedural details have been published elsewhere (Borowicz et al., 1995). Shortly, mice avoiding the dark compartment for over 60 s (on the day after their entry of this compartment had been punished by an electric footshock of 0.8 mA for 2 s) showed no long-term memory impairment and were regarded as remembering the task. Retention was expressed as the percentage of mice with no memory impairment. Sterile saline was used to bring CGP 40116 and CGP 43487 into solution. |
| References |
[1]. Ca2+ channel blockade and the antielectroshock activity of NMDA receptor antagonists, CGP 40116 and CGP 43487, in mice. Eur J Pharmacol. 1996 Sep 19;312(1):27-33. [2]. The competitive NMDA antagonists CGP 43487 and APV potentiate dopaminergic function. Psychopharmacology (Berl). 1995 Apr;118(3):310-5. |
| Additional Infomation |
It has been shown that the Ca 2+ channel agonist, BAY k-8644, exclusively impairs the anticonvulsant action of competitive NMDA receptor antagonists (CGP 37849 and o-CPP-ene) and is completely ineffective in the case of non-competitive NMDA (MK-801) and non-NMDA (NBQX and GYKI 52466) receptor antagonists against electroconvulsions in mice (Czuczwar et al., 1994). These findings could suggest that the potentiation of the anticonvulsant efficacy of CGP 40116 and CGP 43487 was due to the centrally mediated actions of Ca 2+ channel inhibitors. Similarly, the anticonvulsive efficacy of conventional antiepileptics was enhanced by centrally active Ca 2+ channel inhibitors, but not by verapamil (Czuczwar et at., 1990a,b) whose penetration into the brain is limited (Hamann et al., 1983). Epileptiform activity is a sequence of events which may be triggered by excitatory amino acids (Hayashi, t954; Bradford and Peterson, 1987; Meldrum, 1991). First, L-glutamate activates the NMDA receptor/ channel complex, which results in Ca 2 ÷ ion entry through receptor-operated channels following initial depolarization (Mayer and Miller, 1990). Further, the NMDA-activated depolarization spreads to the voltage-operated Ca 2÷ channels and, finally, leads to their activation and a subsequent large Ca z+ influx (Courtney et al., 1990; Mayer and Miller, 1990). Therefore, NMDA receptor antagonists may attenuate Ca 2+ influx either directly through NMDA receptors or indirectly via voltage-operated Ca 2÷ channels. By analogy, similar 'cross-talk' may be expected in the case of Ca 2÷ channel inhibitors. This interpretation might explain the anticonvulsive activity of Ca 2÷ channel inhibitors per se and in combination with CGP 40116 or CGP 43487. There is also evidence that some Ca 2÷ channel inhibitors directly bind to the NMDA receptor complex and produce a specific block of its function (Hashim et al., 1988; Skeen et al., 1993). However, with only few exceptions, there is little experimental support for this hypothesis. Moreover, the presynaptic prevention of excitatory amino acid release by Ca 2+ channel inhibitors cannot be completely ruled out (De Sarro et al., 1988; Janis and Triggle, 1991). The fact that nicardipine, nifedipine and flunarizine enhanced the anticonvulsant action of CGP 40116 and CGP 43487 in a similar fashion may point out that it is Ca 2+ channel blockade which is responsible for this effect. Flunarizine, apart from this mechanism of action, may also block sodium conductances (Ashton and Wauquier, 1986) and this effect could be also involved. Unfortunately, the potential antiepileptic utility of NMDA receptor antagonists is limited by their side-effects (Schmidt, 1994; Witkin, 1995). In fact, the marked potentiation of the protective action of CGP 40116 or CGP 43487 by Ca 2 ÷ inhibitors was accompanied by behavioral impairment. However, the novel strategy (Ca 2÷ channel inhibitors + NMDA receptor antagonists) for the treatment of epilepsy should not be immediately discarded since more specific and less toxic compounds may be soon available. Whether such a polypharmacological approach might be useful in the treatment of other neurological disorders is a question for further studies. [1] According to other studies (Liljequist 1991; Ornstein et al. 1987; Starr and Starr 1994a,b), while MK-801 induced marked hyperactivity at low doses, no dose of CGP 43487 and APV increased rat activity. High doses of either non competitive or competitive NMDA antagonists reduced animal spontaneous motility with different behavioral profiles, MK-801 showing flattened posture and hind limb abduction, while non competitive blockers induced only more pronounced sedation. Indeed, the rota-rod test showed that there is no significative difference in motor coordination before and after CGP 43487 treatment, suggesting that competitive antagonists affect animal spontaneous behavior to a lesser extent than the non competitive drugs and that, among the antagonists studied CGP 43487 could have a clear therapeutic advantage. The most interesting results of the present study relate to the actions of NMDA antagonists on behavioral effect induced by dopaminergic stimulation. Either competitive or non competitive NMDA blockers potentiated the response to the mixed D1/D2 agonist apomorphine at doses which do not themselves influence animal spontaneous behavior. Nevertheless, the administration of MK-801, unlike pretreatment with both competitive NMDA antagonists, did not significantly change the frequency of licking response. According to Fray et al. (1980), licking is not evident at high doses of apomorphine, where it is replaced largely by gnawing. This indicates that greater stimulation ofdopamine receptors results preferentially in gnawing and biting. Nevertheless, in our study, MK-801 could not have potentiated apomorphine response so much as to counteract the licking behavior, as even lower doses of the non competitive NMDA antagonist did not increase the frequency of this behavior (data not shown). Moreover, higher doses of NMDA competitive antagonists potentiated the frequency of licking as well as gnawing behavior. The observation that sniffing and locomotion induced by apomorphine were similar in all pretreated groups, whereas NMDA antagonists-treated rats showed increased frequencies of gnawing, could suggest that the dopaminergic potentiation occurs preferentially in the striatum, the brain area mostly involved in apomorphine-induced gnawing (Ernst 1967; Ljungberg and Ungerstedt 1977). This is in accordance with previous studies suggesting that the antagonistic interaction between central glutamatergic and dopaminergic systems takes place within the striatum (Carlsson and Carlsson 1989; Carlsson and Svensson 1990). Nevertheless, our data do not fully agree with other findings showing that in monoamine-depleted mice, NMDA receptor blockade facilitates the D1 mediated locomotor responses, while it does not affect the behaviors evoked by DI/D2 dopamine receptor stimulation (Verma and Kulkarni 1992; Starr and Starr 1993). The administration of NMDA antagonists to 314 monoamine-depleted animals could reveal the activational potential of other transmitter systems (Carlsson and Carlsson 1989; Carlsson and Svensson 1990). The different experimental conditions (naive versus monoamine-depleted animals) and species could account for our results concerning the potentiating effect of NMDA antagonists on responses evoked by D1/D2 stimulation. In conclusion, the present results agree with other data showing a thcilitating effect of dopamine-mediated behaviors by drugs which block NMDA receptor complex, the competitive NMDA antagonists appearing less likely to cause motor impairment and psychostimulation. [2] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.2160 mL | 21.0801 mL | 42.1603 mL | |
| 5 mM | 0.8432 mL | 4.2160 mL | 8.4321 mL | |
| 10 mM | 0.4216 mL | 2.1080 mL | 4.2160 mL |