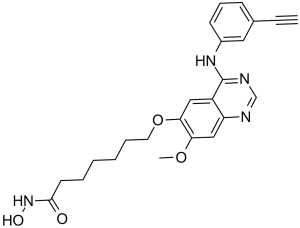
CUDC-101 is a novel, potent and multi-targeted histone deacetylase (HDAC) inhibitor with potential anticancer activity. Moreover, it has EGFR and HER2 inhibition at IC50 values of 2.4 nM, 15.7 nM, and 4.4 nM.
Physicochemical Properties
| Molecular Formula | C24H26N4O4 | |
| Molecular Weight | 434.49 | |
| Exact Mass | 434.195 | |
| Elemental Analysis | C, 66.34; H, 6.03; N, 12.89; O, 14.73 | |
| CAS # | 1012054-59-9 | |
| Related CAS # |
|
|
| PubChem CID | 24756910 | |
| Appearance | White to yellow solid powder | |
| Density | 1.3±0.1 g/cm3 | |
| Melting Point | 174-177ºC | |
| Index of Refraction | 1.638 | |
| LogP | 2.84 | |
| Hydrogen Bond Donor Count | 3 | |
| Hydrogen Bond Acceptor Count | 7 | |
| Rotatable Bond Count | 12 | |
| Heavy Atom Count | 32 | |
| Complexity | 624 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | O(C1=C(C([H])=C2C(C(=NC([H])=N2)N([H])C2=C([H])C([H])=C([H])C(C#C[H])=C2[H])=C1[H])OC([H])([H])[H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C(N([H])O[H])=O |
|
| InChi Key | PLIVFNIUGLLCEK-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C24H26N4O4/c1-3-17-9-8-10-18(13-17)27-24-19-14-22(21(31-2)15-20(19)25-16-26-24)32-12-7-5-4-6-11-23(29)28-30/h1,8-10,13-16,30H,4-7,11-12H2,2H3,(H,28,29)(H,25,26,27) | |
| Chemical Name | 7-[4-(3-ethynylanilino)-7-methoxyquinazolin-6-yl]oxy-N-hydroxyheptanamide | |
| Synonyms | CUDC-101; CUDC 101; 7-(4-(3-Ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide; CUDC101; 7-[[4-(3-Ethynylphenylamino)-7-methoxyquinazolin-6-yl]oxy]-N-hydroxyheptanamide; CHEMBL598797; 1A7Y9MP123; CUDC101 | |
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | EGFR ( IC50 = 2.4 nM ); HER2 ( IC50 = 15.7 nM ); HDAC ( IC50 = 4.4 nM ); HDAC1 ( IC50 = 4.5 nM ); HDAC2 ( IC50 = 12.6 nM ); HDAC3 ( IC50 = 13.2 nM ); HDAC6 ( IC50 = 5.1 nM ); HDAC5 ( IC50 = 11.4 nM ); HDAC9 ( IC50 = 67.2 nM ); HDAC10 ( IC50 = 26.1 nM ); HDAC8 ( IC50 = 79.8 nM ); HDAC7 ( IC50 = 373 nM ) | |
| ln Vitro |
|
|
| ln Vivo |
|
|
| Enzyme Assay | The Biomol Color de Lys method is used to evaluate the actions of Class I and II HDACs. In a nutshell, HDACs are obtained from nuclear extracts from HeLa cells. HeLa cell nuclear extracts are treated with varying concentrations of CUDC-101 in the presence of an artificial colorimetric substrate. Enzyme activity is measured in the Wallac Victor II 1420 microplate reader at 405 nM after developer is added at the conclusion of the assay. HTScan EGF receptor and HER2 kinase assay kits are used to measure EGFR and HER2 kinase activity. In brief, 400 mM ATP is added to an incubation mixture containing different concentrations of CUDC-101 and synthetic biotinylated peptide substrate for the GST-EGFR fusion protein. Strapavidin-coated 96-well plates are used to capture phosphorylated substrate. Secondary antibodies labeled with antiphospho-tyrosine and europium measure the amount of phosphorylation. At the conclusion of the experiment, the enhancement solution is added, and the Wallac Victor II 1420 microplate reader is used to measure the enzyme activity at 615 nM. | |
| Cell Assay | In 96-well flatbottomed plates, cancer cell lines are plated at 5000–10,000 cells per well at different CUDC–101 concentrations. For 72 hours, CUDC-101 is incubated with the cells in the presence of 0.5% fetal bovine serum. Using the Perkin-Elmer ATPlite kit, an adenosine triphosphate (ATP) content assay is used to evaluate growth inhibition. The Apo-ONE Homogeneous Assay Kit is used to measure the activities of Caspase-3 and -7 in order to routinely assess apoptosis. | |
| Animal Protocol |
|
|
| References |
[1]. Discovery of 7-(4-(3-Ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a Potent Multi-Acting HDAC, EGFR, and HER2 Inhibitor for the Treatment of Cancer J. Med. Chem., 2010, 53 (5), pp 2000–2009. [2]. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity.Cancer Res. 2010 May 1;70(9):3647-56. Epub 2010 Apr 13. [3]. CUDC-101, a Novel Inhibitor of Full-Length Androgen Receptor (flAR) and Androgen Receptor Variant 7 (AR-V7) Activity: Mechanism of Action and In Vivo Efficacy. Horm Cancer. 2016 Jun;7(3):196-210. [4]. Dual inhibition of HDAC and EGFR signaling with CUDC-101 induces potent suppression of tumor growth and metastasis in anaplastic thyroid cancer. Oncotarget. 2015 Apr 20;6(11):9073-85. |
|
| Additional Infomation |
CUDC-101 has been used in trials studying the treatment of Cancer, Tumors, Liver Cancer, Breast Cancer, and Gastric Cancer, among others. HDAC/EGFR/HER2 Inhibitor CUDC-101 is a multi-targeted, small-molecule inhibitor of histone deacetylase (HDAC), epidermal growth factor receptor tyrosine kinase (EGFR/ErbB1), and human epidermal growth factor receptor 2 tyrosine kinase (HER2/neu or ErbB2) with potential antineoplastic activity. HDAC/EGFR/HER2 inhibitor CUDC-101 inhibits the activity of these three enzymes but the exact mechanism of action is presently unknown. This agent may help overcome resistance to inhibition of EGFR and Her2 through a simultaneous, synergistic inhibition of EGFR, Her2, and HDAC. By incorporating histone deacetylase (HDAC) inhibitory functionality into the pharmacophore of the epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) inhibitors, we synthesized a novel series of compounds with potent, multiacting HDAC, EGFR, and HER2 inhibition and identified 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide 8 (CUDC-101) as a drug candidate, which is now in clinical development. 8 displays potent in vitro inhibitory activity against HDAC, EGFR, and HER2 with an IC(50) of 4.4, 2.4, and 15.7 nM, respectively. In most tumor cell lines tested, 8 exhibits efficient antiproliferative activity with greater potency than vorinostat (SAHA), erlotinib, lapatinib, and combinations of vorinostat/erlotinib and vorinostat/lapatinib. In vivo, 8 promotes tumor regression or inhibition in various cancer xenograft models including nonsmall cell lung cancer (NSCLC), liver, breast, head and neck, colon, and pancreatic cancers. These results suggest that a single compound that simultaneously inhibits HDAC, EGFR, and HER2 may offer greater therapeutic benefits in cancer over single-acting agents through the interference with multiple pathways and potential synergy among HDAC and EGFR/HER2 inhibitors.[1] Receptor tyrosine kinase inhibitors have recently become important therapeutics for a variety of cancers. However, due to the heterogeneous and dynamic nature of tumors, the effectiveness of these agents is often hindered by poor response rates and acquired drug resistance. To overcome these limitations, we created a novel small molecule, CUDC-101, which simultaneously inhibits histone deacetylase and the receptor kinases epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) in cancer cells. Because of its integrated histone deacetylase inhibition, CUDC-101 synergistically blocked key regulators of EGFR/HER2 signaling pathways, also attenuating multiple compensatory pathways, such as AKT, HER3, and MET, which enable cancer cells to escape the effects of conventional EGFR/HER2 inhibitors. CUDC-101 displayed potent antiproliferative and proapoptotic activities against cultured and implanted tumor cells that are sensitive or resistant to several approved single-targeted drugs. Our results show that CUDC-101 has the potential to dramatically improve the treatment of heterogeneous and drug-resistant tumors that cannot be controlled with single-target agents. Further, they provide a framework to create individual small molecules that simultaneously antagonize multiple biochemically distinct oncogenic targets, suggesting a general paradigm to surpass conventional, single-target cancer therapeutics. [2] Castration-resistant prostate cancer (CRPC) is an androgen receptor (AR)-dependent disease expected to cause the death of more than 27,000 Americans in 2015. There are only a few available treatments for CRPC, making the discovery of new drugs an urgent need. We report that CUDC-101 (an inhibitor od HER2/NEU, EGFR and HDAC) inhibits both the full length AR (flAR) and the AR variant AR-V7. This observation prompted experiments to discover which of the known activities of CUDC-101 is responsible for the inhibition of flAR/AR-V7 signaling. We used pharmacologic and genetic approaches, and found that the effect of CUDC-101 on flAR and AR-V7 was duplicated only by other HDAC inhibitors, or by silencing the HDAC isoforms HDAC5 and HDAC10. We observed that CUDC-101 treatment or AR-V7 silencing by RNAi equally reduced transcription of the AR-V7 target gene, PSA, without affecting viability of 22Rv1 cells. However, when cellular proliferation was used as an end point, CUDC-101 was more effective than AR-V7 silencing, raising the prospect that CUDC-101 has additional targets beside AR-V7. In support of this, we found that CUDC-101 increased the expression of the cyclin-dependent kinase inhibitor p21, and decreased that of the oncogene HER2/NEU. To determine if CUDC-101 reduces growth in a xenograft model of prostate cancer, this drug was given for 14 days to castrated male SCID mice inoculated with 22Rv1 cells. Compared to vehicle, CUDC-101 reduced xenograft growth in a statistically significant way, and without macroscopic side effects. These studies demonstrate that CUDC-101 inhibits wtAR and AR-V7 activity and growth of 22Rv1 cells in vitro and in vivo. These effects result from the ability of CUDC-101 to target not only HDAC signaling, which was associated with decreased flAR and AR-V7 activity, but multiple additional oncogenic pathways. These observations raise the possibility that treatment of CRPC may be achieved by using similarly multi-targeted approaches.[3] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (4.79 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (4.79 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.08 mg/mL (4.79 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 15% Captisol: 30mg/mL Solubility in Formulation 5: 16.67 mg/mL (38.37 mM) in 50% PEG300 50% Saline (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3015 mL | 11.5077 mL | 23.0155 mL | |
| 5 mM | 0.4603 mL | 2.3015 mL | 4.6031 mL | |
| 10 mM | 0.2302 mL | 1.1508 mL | 2.3015 mL |