Physicochemical Properties
| Molecular Formula | C23H25N3O5S |
| Molecular Weight | 455.526704549789 |
| Exact Mass | 455.151 |
| CAS # | 1379445-54-1 |
| PubChem CID | 67607161 |
| Appearance | Typically exists as solid at room temperature |
| LogP | 2.1 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 7 |
| Heavy Atom Count | 32 |
| Complexity | 730 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | NAFLVUYNUAWSOI-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C23H25N3O5S/c1-17-20(15-22(27)28)23(18-7-3-2-4-8-18)24-26(17)16-19-9-5-6-10-21(19)32(29,30)25-11-13-31-14-12-25/h2-10H,11-16H2,1H3,(H,27,28) |
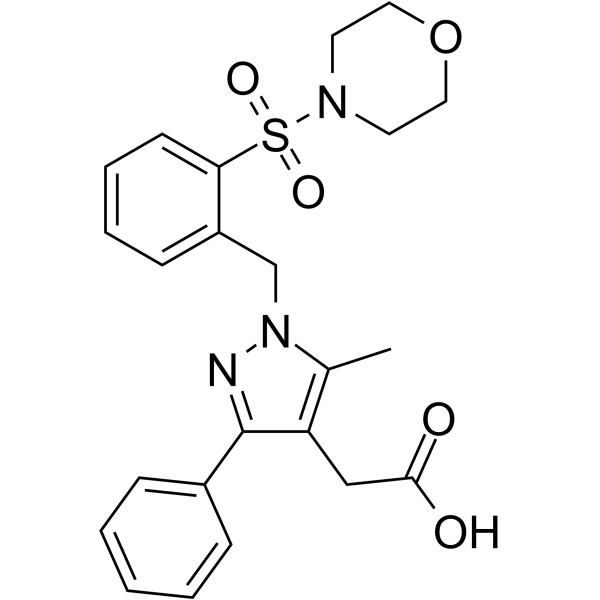
| Chemical Name | 2-[5-methyl-1-[(2-morpholin-4-ylsulfonylphenyl)methyl]-3-phenylpyrazol-4-yl]acetic acid |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | The compounds described are antagonists of the Chemoattractant Receptor-homologous molecule expressed on Th2 cells (CRTh2, also known as DP2) receptor, a Gi-coupled GPCR for prostaglandin D2 (PGD2). The reported activities are primarily IC50 values from functional or binding assays . They are selective over the DP1 and thromboxane A2 receptors (<40% inhibition at 10 µM for tested compounds).[1] |
| ln Vitro |
CRTh2 antagonist 1 is compound 32 [1]. The initial hit compound 5 (structure not fully detailed in main text) was identified from HTS as a micromolar CRTh2 antagonist.[1] Compound 6 (a dimethyl analogue of the hit) showed improved activity, blocking radiolabelled [³⁵S]-GTPγS binding to CRTh2 with an IC50 in a functional assay (exact value not provided, described as more active than hit 5). In a functional eosinophil shape change (ESC) assay in human whole blood (hWB), it showed activity albeit with a 10-fold drop in potency compared to the binding assay.[1] Compound 25 (a pyrazole-4-acetic acid with an ortho-sulfonylbenzyl tail) is a very potent CRTh2 antagonist with an IC50 of 7 nM in the [³⁵S]-GTPγS binding assay. However, its potency significantly shifted in the presence of plasma proteins, showing an IC50 of 2400 nM in the human whole blood ESC assay.[1] Compound 39 (featuring a 4-pyridyl group at the pyrazole 3-position) maintained high potency with an IC50 of 4 nM in the [³⁵S]-GTPγS binding assay and an IC50 of 86 nM in a surrogate assay (GTPγS binding in the presence of 1% human serum albumin), indicating reduced impact of protein binding.[1] Compound 47 (a benzhydryl tail analogue with a 4-pyridyl group) showed an IC50 of 46 nM in the GTPγS binding assay.[1] Compounds 24, 25, 27, 30, 32, 40, and 47 were tested for selectivity against the DP1 and thromboxane A2 (TP) receptors and showed <40% inhibition at 10 µM, indicating good selectivity.[1] Dissociation kinetics were measured for some compounds. Benzyl-substituted compounds 24 and 25 showed rapid dissociation with half-lives of 2 and 12 minutes, respectively. Benzhydryl analogue 47 had a slower dissociation half-life of about 30 minutes.[1] |
| Enzyme Assay | The primary assay used to evaluate CRTh2 antagonism was a [³⁵S]-GTPγS binding assay. Membranes from CHO-K1 cells stably overexpressing the human CRTh2 receptor were pre-incubated with test compounds for 1 hour. Subsequently, the assay was initiated by adding PGD2 (agonist, 50 nM) and a low concentration of [³⁵S]-GTPγS (0.1 nM) in incubation buffer containing MgCl2, NaCl, GDP, and saponin, with either 0.2% bovine serum albumin (BSA) or 1% human serum albumin (HSA) as appropriate. The reaction proceeded for 2 hours at room temperature before termination by filtration. Bound radioactivity was quantified using a liquid scintillation counter. IC50 values were determined from the reduction of signal compared to agonist control.[1] |
| Cell Assay | A functional eosinophil shape change (ESC) assay in human whole blood was employed. Fresh heparinized human whole blood was incubated with test compounds for 10 minutes at room temperature. PGD2 was then added to stimulate the cells for 4 minutes at 37°C. The reaction was stopped on ice, and red blood cells were lysed using an ammonium chloride-based lysis buffer. Remaining cells were fixed and analyzed by flow cytometry. Eosinophils were gated based on autofluorescence, and the change in cell shape (a measure of activation) was assessed by analyzing forward scatter and side scatter profiles. Compound IC50 values were calculated from the inhibition of this shape change response.[1] |
| Animal Protocol |
The literature describes a pharmacokinetic study in rats for compound 6 and 25, but not a specific efficacy study protocol.[1] For compound 6, intravenous (IV) and oral (PO) dosing was performed in rats to assess basic PK parameters. Specific doses, formulation, and frequency are not detailed in the provided text.[1] For compound 25, an IV pharmacokinetic study was conducted in rats at a dose of 1 mg/kg. An oral study was also performed at a dose of 10 mg/kg. The formulation details are not provided.[1] |
| ADME/Pharmacokinetics |
For hit compound 6 in rats: It showed low clearance and good exposure after intravenous dosing. Oral bioavailability was low (exact value not provided).[1] For optimized compound 25 in rats (IV dose 1 mg/kg): AUC0-∞ was 1955 ng·h/mL, clearance was 8.5 mL/min/kg, terminal half-life (t1/2) was 4.8 hours, volume of distribution at steady state (Vss) was 1.6 L/kg. After an oral dose of 10 mg/kg, the oral bioavailability (F) was 73%.[1] Compound 25 showed good Caco-2 cell permeability (Papp ~13-17 x 10⁻⁶ cm/s) and acceptable solubility at pH 7.4 (0.95 mg/mL), but low solubility at pH 1 (0.003 mg/mL). Its pKa was 4.2 and logD7.4 was 0.7.[1] Plasma protein binding (PPB) for compound 25 was extremely high: 98.8% in rat plasma and 99.9% in human plasma as measured by rapid equilibrium dialysis.[1] |
| Toxicity/Toxicokinetics |
The main toxicity-related concern highlighted for the series, exemplified by compound 25, is extremely high plasma protein binding (99.9% in human), which could limit free drug concentration and efficacy despite good overall PK exposure.[1] |
| References |
[1]. 2-(1H-Pyrazol-4-yl)acetic acids as CRTh2 antagonists. Bioorg Med Chem Lett. 2013 Jun 1;23(11):3349-53. |
| Additional Infomation |
The chemical series is based on a 2-(1H-pyrazol-4-yl)acetic acid core, conceived as a ring-expansion analogue of known indoleacetic acid-based CRTh2 antagonists like indomethacin.[1] Structure-activity relationship (SAR) studies revealed a very tight SAR. Potency was highly sensitive to the structure of the “tail” group. An ortho-sulfonyl linker connecting two aromatic rings (as in compound 25) was optimal, likely due to the specific orthogonal conformation it imposes. Replacing this with more planar linkers (e.g., carbonyl, direct bond) drastically reduced potency.[1] Substitution at the pyrazole 3-position significantly impacted protein binding effects. Lipophilic groups (e.g., phenyl) led to huge potency shifts between buffer and protein-containing assays, while a polar 4-pyridyl group (as in compound 39) helped maintain potency in the presence of albumin.[1] The compounds are intended as potential therapeutic agents for allergic inflammatory diseases by blocking the pro-inflammatory effects of PGD2 mediated through CRTh2 on Th2 lymphocytes, eosinophils, and basophils.[1] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1952 mL | 10.9762 mL | 21.9525 mL | |
| 5 mM | 0.4390 mL | 2.1952 mL | 4.3905 mL | |
| 10 mM | 0.2195 mL | 1.0976 mL | 2.1952 mL |