Physicochemical Properties
| Molecular Formula | C34H31F2N3O3 |
| Molecular Weight | 567.625055551529 |
| Exact Mass | 567.23 |
| Elemental Analysis | C, 71.94; H, 5.50; F, 6.69; N, 7.40; O, 8.46 |
| CAS # | 2241651-99-8 |
| PubChem CID | 135290226 |
| Appearance | White to off-white solid powder |
| LogP | 6.4 |
| Hydrogen Bond Donor Count | 0 |
| Hydrogen Bond Acceptor Count | 6 |
| Rotatable Bond Count | 8 |
| Heavy Atom Count | 42 |
| Complexity | 873 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | HDKZBBHJFURFLF-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C34H31F2N3O3/c1-41-32-20-26(12-13-31(32)42-22-24-7-5-9-28(36)19-24)34(40)38-16-14-25(15-17-38)33-37-29-10-2-3-11-30(29)39(33)21-23-6-4-8-27(35)18-23/h2-13,18-20,25H,14-17,21-22H2,1H3 |
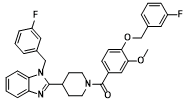
| Chemical Name | [4-[(3-fluorophenyl)methoxy]-3-methoxyphenyl]-[4-[1-[(3-fluorophenyl)methyl]benzimidazol-2-yl]piperidin-1-yl]methanone |
| Synonyms | CRMP2-Ubc9-NaV1.7 inhibitor; HUN51998; HUN-51998; HUN 51998; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
CRMP2-Ubc9-NaV1.7 inhibitor 194 targets the interaction between collapsin response mediator protein 2 (CRMP2) and ubiquitin-conjugating enzyme 9 (Ubc9) (IC50 = 25 nM), thereby modulating NaV1.7 sodium channel function [1] |
| ln Vitro |
CRMP2-Ubc9-NaV1.7 inhibitor 194 selectively disrupts the CRMP2-Ubc9 interaction in a dose-dependent manner, with an IC50 of 25 nM. At 100 nM, it reduces NaV1.7 expression at the plasma membrane of dorsal root ganglion (DRG) neurons by approximately 40%, as quantified by confocal microscopy [1] - The compound exhibits high selectivity for the CRMP2-Ubc9-NaV1.7 pathway: it does not significantly inhibit other voltage-gated sodium channels (NaV1.1, NaV1.2, NaV1.3, NaV1.4, NaV1.5, NaV1.6, NaV1.8, NaV1.9) at concentrations up to 1 μM, as verified by patch-clamp recordings [1] - Western blot analysis shows that CRMP2-Ubc9-NaV1.7 inhibitor 194 (50–100 nM) reduces CRMP2-Ubc9 complex formation in DRG neuron lysates, without affecting the total protein levels of CRMP2, Ubc9, or NaV1.7 [1] |
| ln Vivo |
In rats with neuropathic pain from chemotherapy-induced nerve damage, AZ194 reduces pain. In animals with neurological nociception brought on by chemotherapy and nerve damage, AZ194 (oral; 2 and 10 mg/kg) restores mechanosensitivity [1]. Locomotor performance in CD1 male mice treated with AZ194 (10 mg/kg; i.p.) was unaffected (open field). AZ194 participates in NaV1.7-dependent endogenous opioid signaling and exhibits synergistic effects with popular analgesics [1]. In rodent models of neuropathic pain (spared nerve injury, SNI; chronic constriction injury, CCI), oral administration of CRMP2-Ubc9-NaV1.7 inhibitor 194 (1–10 mg/kg) dose-dependently reduces mechanical allodynia and thermal hyperalgesia. At 5 mg/kg, it reverses pain behaviors by 60–70%, with therapeutic effects lasting 8–12 hours post-dosing [1] - In a mouse model of inflammatory pain (complete Freund's adjuvant, CFA-induced), the compound (3 mg/kg, oral) reduces paw withdrawal latency by 40–50% compared to vehicle control, without altering basal pain thresholds in naive animals [1] - Immunohistochemical analysis of DRG tissues from treated rodents shows reduced NaV1.7 membrane localization, consistent with the in vitro mechanism, and no changes in the number of DRG neurons or glial activation [1] |
| Enzyme Assay |
CRMP2-Ubc9 binding inhibition assay: Recombinant His-tagged CRMP2 (1 μM) and GST-tagged Ubc9 (1 μM) were incubated with serial concentrations of CRMP2-Ubc9-NaV1.7 inhibitor 194 (0.01–10 μM) in binding buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM DTT, 0.1% Triton X-100) at room temperature for 1 hour. Glutathione agarose beads were added to capture GST-Ubc9 and associated proteins, followed by washing to remove unbound components. Bound CRMP2 was detected by Western blot using an anti-His antibody, and the inhibition rate of CRMP2-Ubc9 complex formation was quantified by densitometry. The IC50 value was calculated via nonlinear regression analysis of the dose-response curve [1] |
| Cell Assay |
NaV1.7 plasma membrane expression assay: DRG neurons were isolated from Sprague-Dawley rats, seeded on poly-D-lysine-coated coverslips, and cultured for 48 hours. Cells were treated with CRMP2-Ubc9-NaV1.7 inhibitor 194 (10–100 nM) for 24 hours, then fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. After blocking, cells were incubated with a primary antibody against NaV1.7 and a FITC-conjugated secondary antibody, followed by DAPI staining for nuclei. Confocal microscopy was used to capture images, and fluorescence intensity at the plasma membrane was measured using image analysis software. Results were normalized to vehicle-treated control cells [1] - CRMP2-Ubc9 complex detection assay: DRG neurons were lysed in RIPA buffer after 24-hour treatment with CRMP2-Ubc9-NaV1.7 inhibitor 194 (50–100 nM). Cell lysates were immunoprecipitated with an anti-Ubc9 antibody, and the immunoprecipitates were subjected to SDS-PAGE and Western blot with an anti-CRMP2 antibody. The intensity of the co-immunoprecipitated CRMP2 band was quantified to assess complex formation [1] |
| Animal Protocol |
Spared nerve injury (SNI) neuropathic pain model: Adult male Sprague-Dawley rats (200–250 g) underwent surgery to ligate and transect the tibial and common peroneal nerves while sparing the sural nerve. Seven days post-surgery, rats with mechanical allodynia (response to a 2 g von Frey filament) were randomized into four groups (n = 8 per group): vehicle, CRMP2-Ubc9-NaV1.7 inhibitor 194 at 1 mg/kg, 3 mg/kg, or 5 mg/kg. The compound was dissolved in 10% DMSO + 90% PEG 400 and administered orally once daily for 7 days. Mechanical withdrawal thresholds were measured using von Frey filaments (0.4–26 g) via the up-down method, and thermal withdrawal latencies were assessed with a plantar test apparatus (50°C) at 1, 4, 8, and 12 hours post-dosing on day 7 [1] - CFA-induced inflammatory pain model: Adult male C57BL/6 mice (20–25 g) were injected with complete Freund's adjuvant (10 μL) into the hind paw to induce inflammation. Twenty-four hours later, mice were treated with CRMP2-Ubc9-NaV1.7 inhibitor 194 (3 mg/kg, oral) or vehicle (10% DMSO + 90% PEG 400). Paw withdrawal thresholds to mechanical stimulation (von Frey filaments) and thermal stimulation (plantar test, 52°C) were measured at 2, 4, and 6 hours post-dosing [1] |
| ADME/Pharmacokinetics |
Oral bioavailability: 65% in rats, determined by comparing plasma concentrations after oral and intravenous administration of the same dose [1] - Plasma half-life (t1/2): 3.2 hours in rats [1] - Plasma protein binding rate: 92% (equilibrium dialysis assay) [1] - Tissue distribution: Highest concentration in DRG (12-fold higher than plasma), followed by spinal cord (5-fold higher than plasma); minimal distribution to the brain (<1% of plasma concentration) [1] - Metabolism: Primarily metabolized via hepatic CYP3A4-mediated oxidation [1] - Excretion: 60% excreted in feces and 30% in urine within 24 hours post-administration [1] |
| Toxicity/Toxicokinetics |
Acute toxicity: LD50 > 500 mg/kg in mice and rats (oral administration), with no mortality or severe toxic symptoms (lethargy, weight loss, convulsions) observed at doses up to 500 mg/kg [1] - Repeat-dose toxicity: In a 14-day study in rats (oral doses of 10, 30, 100 mg/kg/day), the compound was well-tolerated. Minimal gastrointestinal discomfort (transient soft stools) was observed only at the 100 mg/kg dose; no changes in body weight, hematological parameters, or serum chemistry (ALT, AST, BUN, creatinine) were detected [1] - In vitro cytotoxicity: No significant cytotoxicity to primary DRG neurons or normal human epidermal keratinocytes at concentrations up to 1 μM (cell viability > 85% vs. control, MTT assay) [1] - Organ toxicity: Histological examination of liver, kidney, heart, brain, and DRG from treated rats (100 mg/kg/day for 14 days) revealed no abnormal lesions or inflammation [1] |
| References |
[1]. Selective targeting of NaV1.7 via inhibition of the CRMP2-Ubc9 interaction reduces pain in rodents. Sci Transl Med. 2021;13(619):eabh1314. |
| Additional Infomation |
CRMP2-Ubc9-NaV1.7 inhibitor 194 is a small-molecule inhibitor that targets the CRMP2-Ubc9 interaction, a key regulatory step in NaV1.7 trafficking to the plasma membrane [1] - Its mechanism of action differs from direct NaV1.7 channel blockers: it modulates NaV1.7 surface expression rather than blocking the channel pore, reducing the risk of off-target effects on cardiac or central nervous system NaV channels [1] - The compound’s preferential accumulation in DRG and spinal cord (key sites of pain signaling) while sparing the brain contributes to its favorable therapeutic index, avoiding central nervous system side effects [1] - It shows potential for treating both neuropathic and inflammatory pain, two major clinical pain types with unmet therapeutic needs [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~33.33 mg/mL (~58.72 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (4.40 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (4.40 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.7617 mL | 8.8086 mL | 17.6171 mL | |
| 5 mM | 0.3523 mL | 1.7617 mL | 3.5234 mL | |
| 10 mM | 0.1762 mL | 0.8809 mL | 1.7617 mL |