Physicochemical Properties
| Molecular Formula | C26H31FN4O4 |
| Molecular Weight | 482.55 |
| Exact Mass | 482.233 |
| Elemental Analysis | C, 64.71; H, 6.48; F, 3.94; N, 11.61; O, 13.26 |
| CAS # | 212790-31-3 |
| PubChem CID | 5311123 |
| Appearance | Typically exists as solid at room temperature |
| LogP | 4.238 |
| Hydrogen Bond Donor Count | 4 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 11 |
| Heavy Atom Count | 35 |
| Complexity | 709 |
| Defined Atom Stereocenter Count | 3 |
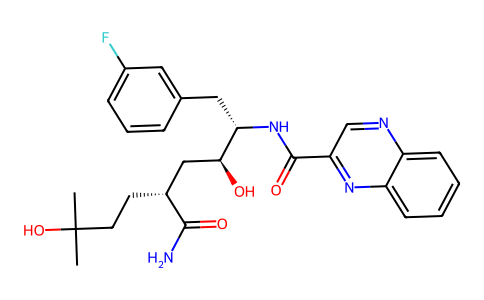
| SMILES | CC(CC[C@@H](C(N)=O)C[C@H](O)[C@@H](NC(C1=NC2=CC=CC=C2N=C1)=O)CC3=CC(F)=CC=C3)(O)C |
| InChi Key | YEQJVHQCUDMXFG-FHZYATBESA-N |
| InChi Code | InChI=1S/C26H31FN4O4/c1-26(2,35)11-10-17(24(28)33)14-23(32)21(13-16-6-5-7-18(27)12-16)31-25(34)22-15-29-19-8-3-4-9-20(19)30-22/h3-9,12,15,17,21,23,32,35H,10-11,13-14H2,1-2H3,(H2,28,33)(H,31,34)/t17-,21+,23+/m1/s1 |
| Chemical Name | N-[(2S,3S,5R)-5-carbamoyl-1-(3-fluorophenyl)-3,8-dihydroxy-8-methylnonan-2-yl]quinoxaline-2-carboxamide |
| Synonyms | CP-481715; CP481715; 212790-31-3; CP-481,715; CP 481715; X3TDA066ME; UNII-X3TDA066ME; N-[(2S,3S,5R)-5-carbamoyl-1-(3-fluorophenyl)-3,8-dihydroxy-8-methylnonan-2-yl]quinoxaline-2-carboxamide; CHEMBL1628706; CP 481715 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | CCR1 (Kd = 9.2 nM) |
| ln Vitro | The chemokines CCL3 and CCL5, as well as their shared receptor CCR1, are believed to play a role in the pathogenesis of several inflammatory diseases including rheumatoid arthritis, multiple sclerosis, and transplant rejection. In this study we describe the pharmacological properties of a novel small molecular weight CCR1 antagonist, CP-481,715 (quinoxaline-2-carboxylic acid [4(R)-carbamoyl-1(S)-(3-fluorobenzyl)-2(S),7-dihydroxy-7-methyloctyl]amide). Radiolabeled binding studies indicate that CP-481,715 binds to human CCR1 with a Kd of 9.2 nm and displaces 125I-labeled CCL3 from CCR1-transfected cells with an IC50 of 74 nm. CP-481,715 lacks intrinsic agonist activity but fully blocks the ability of CCL3 and CCL5 to stimulate receptor signaling (guanosine 5'-O-(thiotriphosphate) incorporation; IC50 = 210 nm), calcium mobilization (IC50 = 71 nm), monocyte chemotaxis (IC50 = 55 nm), and matrix metalloproteinase 9 release (IC50 = 54 nm). CP-481,715 retains activity in human whole blood, inhibiting CCL3-induced CD11b up-regulation and actin polymerization (IC50 = 165 and 57 nm, respectively) on monocytes. Furthermore, it behaves as a competitive and reversible antagonist. CP-481,715 is >100-fold selective for CCR1 as compared with a panel of G-protein-coupled receptors including related chemokine receptors. Evidence for its potential use in human disease is suggested by its ability to inhibit 90% of the monocyte chemotactic activity present in 11/15 rheumatoid arthritis synovial fluid samples. These data illustrate that CP-481,715 is a potent and selective antagonist for CCR1 with therapeutic potential for rheumatoid arthritis and other inflammatory diseases [1]. |
| ln Vivo |
Pretreatment with 1000 mg CP-481715 TID resulted in significant reductions in visual scores of the nickel reactions (P = 0.01). Instrumentally measured erythema tended to decrease in the CP-481715 mg TID group (P = 0.06). No differences were noted between the 3000 mg CP-481715 mg QD group and pooled placebo. No significant differences were found for immunohistological cell counts. CP-418 715 was generally safe and well tolerated.
Conclusions: Blocking of CCR1 only partly inhibited clinical manifestations of ACD. Several chemokine receptors are likely relevant for the cellular influx observed in ACD lesions.[2] We previously described the in vitro characteristics of the potent and selective CCR1 antagonist, CP-481,715. In addition to being selective for CCR1 vs other chemokine receptors, CP-481,715 is also specific for human CCR1 (hCCR1), preventing its evaluation in classical animal models. To address this, we generated mice whereby murine CCR1 was replaced by hCCR1 (knockin) and used these animals to assess the anti-inflammatory properties of CP-481,715. Cells isolated from hCCR1 knockin mice were shown to express hCCR1 and migrate in response to both murine CCR1 and hCCR1 ligands. Furthermore, this migration is inhibited by CP-481,715 at dose levels comparable to those obtained with human cells. In animal models of cell infiltration, CP-481,715 inhibited CCL3-induced neutrophil infiltration into skin or into an air pouch with an ED50 of 0.2 mg/kg. CP-481,715 did not inhibit cell infiltration in wild-type animals expressing murine CCR1. In a more generalized model of inflammation, delayed-type hypersensitivity, CP-481,715 significantly inhibited footpad swelling and decreased the amount of IFN-gamma and IL-2 produced by isolated spleen cells from sensitized animals. It did not, however, induce tolerance to a subsequent challenge. These studies illustrate the utility of hCCR1 knockin animals to assess the activity of human specific CCR1 antagonists; demonstrate the ability of the CCR1 antagonist CP-481,715 to inhibit cell infiltration, inflammation, and Th1 cytokine responses in these animals; and suggest that CP-481,715 may be useful to modulate inflammatory responses in human disease[3]. |
| Cell Assay |
Chemotaxis assays [2] Chemotaxis was conducted in 48-well chemotaxis chambers purchased from NeuroProbe, as previously described. Briefly, agonists were diluted in RPMI 1640 containing 0.1% BSA, then added to the bottom wells of the chamber. A polyvinylpyrrolidone-free filter with 5-μm pores was placed between the upper and lower wells of the chamber. Cells were then added to the top chamber (2 × 105) in the presence or absence of various concentrations of CP-481,715, and the apparatus was incubated for 60 min in a 5% CO2 humidified incubator at 37°C. After the incubation period, the nonmigrating cells were removed from the upper chamber, and the top of the filter was wiped. The bottom portion of the filter was stained with Diff-Quik, and the number of migrating cells in six random fields was enumerated with a microscope. Whole blood actin polymerization [2] Mouse blood, collected in EDTA, was incubated with various dilutions of CP-481,715 or diluent for 5 min at room temperature. CCL3 (10 nM) was then added, and after 50 s the reaction was terminated by adding FACS lysing solution containing paraformaldehyde. After 10 min, the cells were collected by centrifugation, washed with PBS, and stained for 1 h at room temperature in the dark with a solution containing lysophosphatidylcholine, paraformaldehyde, and nitrobenzoxadiazole phallacidin. The cells were then washed with PBS containing 2% FBS, and the fluorescence was quantitated using a FACScan |
| Animal Protocol |
Air pouch model of cell infiltration [2] Subcutaneous air pouches were formed on the back of animals, as previously described. Briefly, 3 ml of air was injected s.c. on day 1 and then reinjected again 3 days later in the same area. On the fourth day, animals received a single i.p. injection of CP-481,715, followed by two injections of CCL3 (1 μg/ml) administered directly into the air pouch at time 0 and 2 h. The pouches were washed with 3 ml of PBS containing 10 mM EDTA 2 h after the last injection of CCL3. The number of cells was counted microscopically. Delayed-type hypersensitivity model [2] Delayed-type hypersensitivity was assessed in SRBC-sensitized mice. Briefly, defibrinated SRBC (REMEL) were washed, and 1 × 106 cells were injected i.v. into animals to sensitize them. Six days later, mice were injected into the footpad with 108 SRBCs in 25 μl. Footpad swelling was measured with calipers 24 h after rechallenge. In some animals, CP-481,715 was administered as a single injection (i.p.) at the time of rechallenge. In other studies, CP-481,715 was administered i.p. daily beginning at the time of sensitization. |
| References |
[1]. CP-481,715, a potent and selective CCR1 antagonist with potential therapeutic implications for inflammatory diseases.J Biol Chem. 2003 Oct 17;278(42):40473-80. [2]. Evaluation of the effect of the specific CCR1 antagonist CP-481715 on the clinical and cellular responses observed following epicutaneous nickel challenge in human subjects.Contact Dermatitis. 2008 Oct;59(4):212-9. [3]. The human specific CCR1 antagonist CP-481,715 inhibits cell infiltration and inflammatory responses in human CCR1 transgenic mice.J Immunol. 2006 Mar 1;176(5):3141-8. |
| Additional Infomation |
Background: The CC-chemokine receptor-1 (CCR1) is thought to be involved in recruitment of inflammatory cells in allergic contact dermatitis (ACD). CP-481715 is a specific antagonist of CCR1.
Objectives: To determine the inhibitory effects of CP-418 715 in ACD by evaluating the clinical signs and cellular infiltration in skin biopsies following epicutaneous nickel challenge in allergic subjects.
Subjects/methods: In this phase 1/2 study, 40 subjects were randomized to 5 days of treatment in four parallel groups (placebo three times daily (TID), placebo once daily (QD), 1000 mg CP-418 715 TID, and 3000 mg CP-418 715 QD). Twenty-four hours after the first drug administration, nickel sulfate patches were applied on subjects' backs and removed 48 hours later.[2] Generation of human chemokine receptor KI animals represents a viable strategy to assess the in vivo activity of human specific chemokine receptor antagonists. Leukocytes from KI animals express hCCR1 and migrate to CCR1 ligands. Studies in hCCR1 KI animals demonstrate the potent ability of CP-481,715 to decrease CCL3-induced cell infiltration, prevent inflammatory responses (delayed-type hypersensitivity), and alter cytokine responses in sensitized animals. These studies underscore the importance of CCR1 in inflammation and the role of chemokines in these responses, and raise the possibility that inhibiting CCR1 will modulate inflammatory responses in clinic. In addition, the ability of CP-481,715 to inhibit cell infiltration at dose levels and plasma concentrations achievable in clinic suggests the potential clinical utility of this agent in human inflammatory diseases.[2] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0723 mL | 10.3616 mL | 20.7232 mL | |
| 5 mM | 0.4145 mL | 2.0723 mL | 4.1446 mL | |
| 10 mM | 0.2072 mL | 1.0362 mL | 2.0723 mL |