Physicochemical Properties
| Molecular Formula | C16H23N3O2S |
| Molecular Weight | 321.43772 |
| Exact Mass | 321.151 |
| Elemental Analysis | C, 59.79; H, 7.21; N, 13.07; O, 9.95; S, 9.97 |
| CAS # | 143321-74-8 |
| PubChem CID | 132552 |
| Appearance | Typically exists as solid at room temperature |
| LogP | 1.5 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 4 |
| Rotatable Bond Count | 5 |
| Heavy Atom Count | 22 |
| Complexity | 476 |
| Defined Atom Stereocenter Count | 1 |
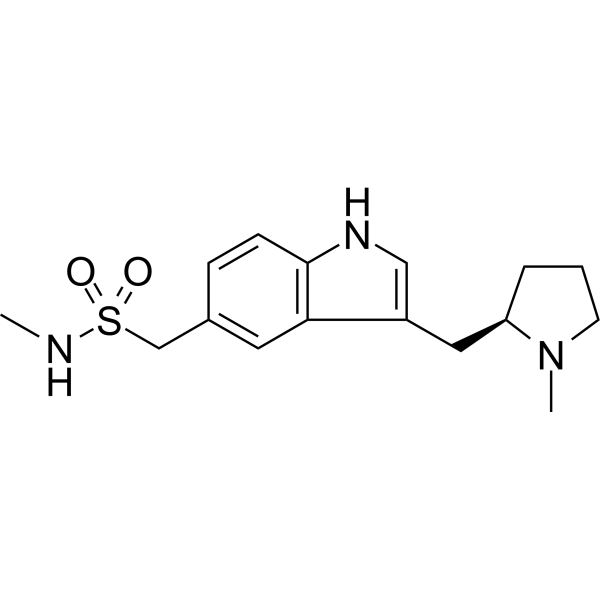
| SMILES | CNS(=O)(=O)CC1=CC2=C(C=C1)NC=C2C[C@H]3CCCN3C |
| InChi Key | BWQZTHPHLITOOZ-CQSZACIVSA-N |
| InChi Code | InChI=1S/C16H23N3O2S/c1-17-22(20,21)11-12-5-6-16-15(8-12)13(10-18-16)9-14-4-3-7-19(14)2/h5-6,8,10,14,17-18H,3-4,7,9,11H2,1-2H3/t14-/m1/s1 |
| Chemical Name | N-methyl-1-[3-[[(2R)-1-methylpyrrolidin-2-yl]methyl]-1H-indol-5-yl]methanesulfonamide |
| Synonyms | CP122288; CP 122288; CP-122288; 143321-74-8; CP 122,288; CP-122,288; 59E9WCT6EM; N-Methyl-3-((1-methylpyrrolidin-2-yl)methyl)-1H-indole-5-methanesulfonamide; N-methyl-1-[3-[[(2R)-1-methylpyrrolidin-2-yl]methyl]-1H-indol-5-yl]methanesulfonamide; UNII-59E9WCT6EM; CP-122288 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | serotonin receptors 5-HT1B, 5-HT1D and 5-HT1F |
| ln Vitro | CP-122288 is a selective agonist for the serotonin receptors 5-HT1B, 5-HT1D and 5-HT1F. CP-122288 is also an inhibitor of neurogenic inflammation and plasma protein extravasation. |
| ln Vivo |
CP-122,288 is a highly potent inhibitor of neurogenic plasma extravasation in animal models at doses without vasoconstrictor effect. Researchers evaluated the acute antimigraine efficacy of intravenous and oral CP-122,288 in two double-blind studies. In a crossover design, patients randomly received 31.25 microg of CP-122,288 intravenously, placebo, or both. In the oral study, patients received placebo or one of four doses of CP-122,288 between 3.125 and 312.5 microg, using a novel "up and down" design for randomization. Both studies were stopped prematurely when target efficacy could not be achieved. Responder rates were 29% for CP-122,288 versus 30% for placebo (difference, -1%; 95% CI, -24-22%; intravenous study) and an overall rate of 25% for CP-122,288 versus 0% for placebo (difference, 25%; 95% CI; 10-40%; oral study). CP-122,288 was not clinically effective at doses and plasma concentrations in excess of those required to inhibit neurogenic plasma extravasation in animals. Neurogenic plasma extravasation is unlikely to play a crucial role in the pathophysiology of migraine headache[1]. We studied the dural plasma protein extravasation response after unilateral electrical stimulation of the trigeminal ganglion in mice lacking serotonin 5-HT1B (5-HT1D beta) receptors by modifying a technique previously described in rats or guinea pigs. We investigated the inhibitory effects of six 5-HT1 receptor agonists in this model: 3-(1,2,5,6-tetrahydropyrid-4-yl)pyrrolo[3,2-b]pyrid-5-one (CP-93,129), sumatriptan, serotonin-5-O-carboxymethyl-glycyl -tyrosinamide (GTI), 5-methylaminosulfonylmethyl-3-(N-methylpyrrolidin-2R -ylmethyl)-1H-indole (CP-122,288), 5-carboxamido-tryptamine (5-CT), and dihydroergotamine. The plasma extravasation response did not differ between wild-type and mutant after vehicle injection. The potency of sumatriptan, CP-122,288, CP-93,129, and 5-CT in wild-type mice was similar to that previously reported for rats. CP-122,288 (1 nmol kg), 5-CT (1 nmol/kg), and dihydroergotamine (72 nmol/kg) inhibited plasma protein extravasation within dura mater after electrical trigeminal ganglion stimulation in both wild-type and knockout mice, which suggests that these agonists act predominantly via receptors other than 5-HT1B. Unlike the wild-type mice, CP-93,129 (1.4 mumol/kg), a specific 5-HT1B receptor agonist, had no effect in knockout mice. The same held true for sumatriptan (0.7 mumol/kg) and GTI (0.6 mumol/kg). These results suggest that CP-93,129, sumatriptan, and GTI exert their effects via 5-HT1B (5-HT1D beta) receptors in mice[2]. The aim of this study was to examine whether GR-127,935, a 5-HT1B/1D receptor antagonist, blocks the inhibitory effects of sumatriptan, CP-122,288 and 5-carboxamidotryptamine (5-CT) on plasma protein extravasation, within guinea pig and rat dura matter, following electric stimulation of the trigeminal ganglion. Binding studies first established that GR-127,935 shows a 500-fold selectivity for 5-HT1D binding sites (labeled by [3H]L-694,247) versus 5-HT1F binding sites (labeled by [3H]sumatriptan in the presence of 50 nM 5-carboxamidotryptamine) in guinea pig forebrain homogenates (pKD +/- SD = 7.0 +/- 0.2 at 5-HT1F sites and 9.7 +/- 0.1 at 5-HT1D sites). In guinea pigs, GR-127,935 showed partial agonist activity and inhibited dural plasma protein extravasation. Increasing doses of GR-127,935 reversed the effect of sumatriptan, but did not affect the action of 5-CT and CP-122,288 (at a dose as high as 2 mumol/kg). Sumatriptan, CP 122,288 and 5-CT dose-responsively inhibited plasma protein extravasation. At a dose of 2 mumol/kg (but not at 0.2 mumol/kg), GR-127,935 right-shifted the dose-response curve of sumatriptan. No significant rightward shift was observed in the dose-response of CP-122,288 and 5-CT. In rats, GR-127,935 did not show any significant partial agonist activity. A dose of 0.2 mumol/kg was sufficient to right-shift the dose-response curve of sumatriptan. These data suggest that sumatriptan inhibits neurogenic inflammation via 5-HT1D alpha receptors in guinea pigs and 5-HT1D beta (5-HT1B) receptors in rats. Additional receptor subtypes are likely to be involved in the inhibition of plasma extravasation by CP-122,288 and 5-CT. Pertussis toxin reduced the inhibitory effects of both sumatriptan and 5-CT, but not of muscimol, known to act at GABAA receptors. These results suggest that 5-CT, as well as sumatriptan, act at a receptor linked to an inhibitory G-protein[3]. |
| Animal Protocol | 1. The aim of the present study was to investigate the in vivo pharmacological profile of CP-122,288, an indole-derivative with a conformationally restricted N-methylpyrrolidinyl basic side chain in the C-3 position. This C-3 substituent structurally differentiates CP-122,288 from the 5-HT1D receptor agonist sumatriptan, which possesses an N,N-dimethylaminoethyl group. [Formula: see text] 2. When administered prior to electrical stimulation of the trigeminal ganglion, CP-122,288 (0.3-300 ng kg-1, i.v.) produced a dose-related inhibition of plasma protein extravasation in rat dura mater (minimum effective dose, MED, 3 ng kg-1 i.v., P < 0.05; maximal inhibition of plasma extravasation at 30 ng kg-1 i.v., P < 0.01). Sumatriptan produced a similar inhibition of plasma leakage in the dura, but at much higher dose levels (MED, 100 micrograms kg-1 i.v., P < 0.05). Thus, CP-122,288 is of the order of 10(4) fold more potent than sumatriptan. 3. At all doses tested, CP-122,288 did not inhibit plasma protein extravasation measured in extracranial tissues such as the lower lip, eyelid, and conjunctiva. 4. In a separate series of studies in the anaesthetized rat, CP-122,288 (0.003-3 micrograms kg-1 i.v.) produced no change in either heart rate or mean arterial blood pressure, thus demonstrating that doses of CP-122,288 which inhibit plasma protein leakage in rat dura, are devoid of hemodynamic effects. 5. Following a 5 min period of electrical stimulation of the trigeminal ganglion, a 20 min period of sustained neurogenically-driven plasma extravasation, occurring in the absence of electrical stimulation, was initiated. By administration of the compound 5 min after completing the phase of electrical stimulation, this protocol permitted the evaluation of the activity of CP-122,288 on an ongoing and established inflammatory event. CP-122,288 (30 and 300 ng kg-1, i.v., P < 0.01 and P < 0.05, respectively) produced a complete inhibition of plasma protein leakage which was consistent with its effects when administered prior to trigeminal ganglion stimulation. 6. In the anaesthetized dog, CP-122,288 and sumatriptan, at 1-300 micrograms kg-1, i.v., produced a dose-dependent reduction in carotid arterial blood flow and coronary arterial diameter. These data demonstrate that sumatriptan inhibits neurogenic inflammation in the rat (MED, 100 micrograms kg-1, i.v.), and produces vasoconstriction in the dog, over a similar dose-range. Interestingly, doses of CP-122,288 that inhibit neurogenic inflammation in rat dura mater (0.3-300 ng kg-1) were demonstrated to be devoid of vasoconstrictor activity in either the carotid or coronary vascular beds of dog. 7. These data demonstrate that in the rat, CP-122,288 is a highly potent and selective inhibitor of neurogenic inflammation in intracranial tissues, at doses which are devoid of vasoconstrictor activity in dog. Potentially, CP-122,288 may be of use for the acute treatment of migraine, without the risk of cardiovascular side-effects.[4] |
| References |
[1]. No acute antimigraine efficacy of CP-122,288, a highly potent inhibitor of neurogenic inflammation: results of two randomized, double-blind, placebo-controlled clinical trials. Ann Neurol. 2000 Feb;47(2):238-41. [2]. 5-Carboxamido-tryptamine, CP-122,288 and dihydroergotamine but not sumatriptan, CP-93,129, and serotonin-5-O-carboxymethyl-glycyl -tyrosinamide block dural plasma protein extravasation in knockout mice that lack 5-hydroxytryptamine1B receptors. Mol Pharmacol. 1996 May;49(5):761-5. [3]. The 5-HT1D receptor antagonist GR-127,935 prevents inhibitory effects of sumatriptan but not CP-122,288 and 5-CT on neurogenic plasma extravasation within guinea pig dura mater. Neuropharmacology. 1997 Jan;36(1):83-91. [4]. The in vivo pharmacological profile of a 5-HT1 receptor agonist, CP-122,288, a selective inhibitor of neurogenic inflammation. Br J Pharmacol. 1995 Nov;116(5):2385-90. |
| Additional Infomation |
The pathophysiological basis for the pain of migraine has been the subject of substantial attention and must include activation of elements of the trigeminal innervation of the cranial vessels, the trigeminovascular system. Recently, consideration of trigeminal-evoked neurogenic plasma protein extravasation (PPE) as a model for the pain has driven the search for compounds with specific anti-extravasation properties. Calcitonin gene-related peptide (CGRP) is a marker for trigeminovascular activation and is released during the headache phase of migraine and cluster headache. CGRP may have a role in migraine through its potent cranial vasodilator effects or by an action on trigeminal nerve activity, both of which are targeted by 5HT(1B/1D)agonist drugs but does not itself produce PPE. It has been suggested that 5HT(1B/1D)agonists may have an anti-migraine effect via inhibition of PPE in the dura mater. Avitriptan and CP122,288 both have strong binding affinities for 5HT(1B/1D)receptors, but only CP122,288 is a potent inhibitor of PPE. In this study we sought to compare the effects of CP122,288 and avitriptan on jugular vein CGRP release after stimulation of the superior sagittal sinus (SSS) in the cat. In eleven anaesthetized cats external jugular vein blood samples were analyzed by radioimmunoassay for CGRP levels in three settings: a) control, b) 1 min after SSS stimulation and c) 1 min after SSS stimulation in presence of drug. Stimulation of the SSS resulted in release of CGRP from the external jugular vein (77+/-1 pmol/L). At a PPE-inhibitory dose in rat (100 ng/kg intravenously) CP122, 288 had no effect on CGRP release (77+/-6 pmol/L) whereas at a clinically relevant dose (50 microgram/kg intravenously) avitriptan blocked CGRP release. This study demonstrates that the potent inhibitor of PPE, CP122, 288, which has been shown in clinical trials to be ineffective in treating acute migraine attacks, had no effect on CGRP release, whereas the effective anti-migraine drug and relatively impotent inhibitor of PPE, avitriptan, blocked CGRP release. These data emphasize the importance of CGRP release and its possible independence from PPE in migraine and more importantly suggest that other non-5HT-based pharmacological targets may account for PPE blockade in animal studies. Neuropeptides. 1999 Feb;33(1):41-6. https://pubmed.ncbi.nlm.nih.gov/10657470/
CP-122,288 is a highly potent inhibitor of neurogenic plasma extravasation in animal models at doses without vasoconstrictor effect. We evaluated the acute antimigraine efficacy of intravenous and oral CP-122,288 in two double-blind studies. In a crossover design, patients randomly received 31.25 microg of CP-122,288 intravenously, placebo, or both. In the oral study, patients received placebo or one of four doses of CP-122,288 between 3.125 and 312.5 microg, using a novel "up and down" design for randomization. Both studies were stopped prematurely when target efficacy could not be achieved. Responder rates were 29% for CP-122,288 versus 30% for placebo (difference, -1%; 95% CI, -24-22%; intravenous study) and an overall rate of 25% for CP-122,288 versus 0% for placebo (difference, 25%; 95% CI; 10-40%; oral study). CP-122,288 was not clinically effective at doses and plasma concentrations in excess of those required to inhibit neurogenic plasma extravasation in animals. Neurogenic plasma extravasation is unlikely to play a crucial role in the pathophysiology of migraine headache.Ann Neurol . 2000 Feb;47(2):238-41. https://pubmed.ncbi.nlm.nih.gov/10665496/ CP122,288, a conformationally restricted analogue of sumatriptan, is a highly potent inhibitor of neurogenic plasma protein extravasation (PPE) in rat and guinea pig at low doses where it has no 5HT1B-mediated vascular actions. We have examined its effect on a model of trigeminovascular nociception to assess the relative importance of vasoconstrictor and serotonin (5HT)(1B/1D) agonist activity to the modulation trigeminal neuronal activation. For comparison to activate relevant 5HT receptors, the clinically effective relatively lipophilic 5HT(1B/1D) agonist eletriptan was studied in parallel. The superior sagittal sinus was isolated in the alpha-chloralose (60 mg/kg, i.p. and 15-20 mg/kg i.v. supplement every 2 h) anaesthetized cat. Animals were prepared for stimulation and then maintained for 24 h before stimulation and perfusion for Fos immunohistochemistry. Stimulation of the superior sagittal sinus (250 micros, 100 V, 0.3 Hz) resulted in Fos expression in cells in the trigeminal nucleus caudalis and superficial laminae of the dorsal horns of C(1-2). Administration of low dose CP122,288 (100 ng/kg) had no effect on Fos expression after sinus stimulation either when administered alone or in combination with mannitol; the latter to ensure access to the trigeminocervical complex. The number of cells in the superficial laminae of the trigeminal nucleus caudalis with stimulation being a median of 50 (quartile range: 47-53) and 48 (35-48) after CP122,288, and after CP122,288 and mannitol 45 (41-53). In comparison, the clinically effective 5HT(1B/1D) agonist, eletriptan, reduced Fos expression in the trigeminocervical complex to a median of 24 (21-33). These data demonstrate that the potent inhibitor of neurogenic plasma protein extravasation (PPE) CP122,288 has no effect on Fos expression in central trigeminal neurons when administered at a dose which blocks PPE in rat and guinea pig, but has no vasoconstrictor 5HT(1B/1D) activity, and while ensuring its access to central trigeminal neurons. The data suggest that activation of the 5HT(1B/1D) receptor is important for the clinical action of this class of compounds and is consistent with the fact the CP122,288 is ineffective in the treatment of the acute attack of migraine. Pain . 1999 Jul;82(1):15-22. https://pubmed.ncbi.nlm.nih.gov/10422655/ |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.1110 mL | 15.5550 mL | 31.1100 mL | |
| 5 mM | 0.6222 mL | 3.1110 mL | 6.2220 mL | |
| 10 mM | 0.3111 mL | 1.5555 mL | 3.1110 mL |