Physicochemical Properties
| Molecular Formula | C10H12N4O4.HCL |
| Molecular Weight | 288.68762 |
| Exact Mass | 288.063 |
| CAS # | 134665-72-8 |
| Related CAS # | CNDAC;135598-68-4 |
| PubChem CID | 3035200 |
| Appearance | White to off-white solid powder |
| Boiling Point | 596.9ºC at 760mmHg |
| Flash Point | 314.8ºC |
| Vapour Pressure | 9.77E-17mmHg at 25°C |
| Hydrogen Bond Donor Count | 4 |
| Hydrogen Bond Acceptor Count | 5 |
| Rotatable Bond Count | 2 |
| Heavy Atom Count | 19 |
| Complexity | 466 |
| Defined Atom Stereocenter Count | 4 |
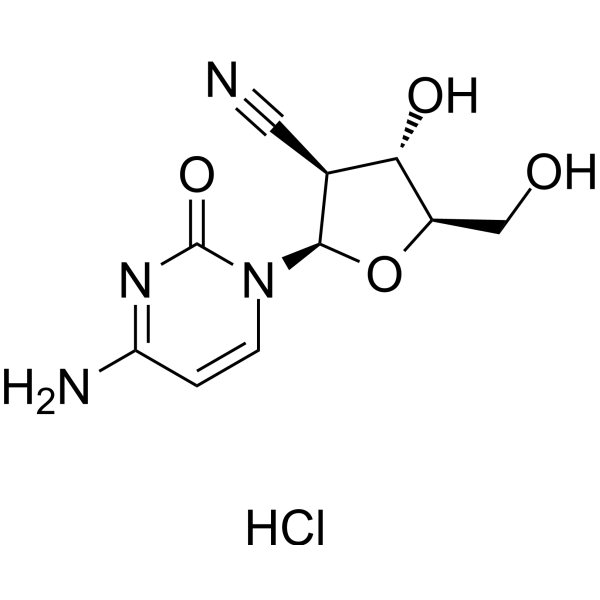
| SMILES | C1=CN(C(=O)N=C1N)[C@H]2[C@H]([C@@H]([C@H](O2)CO)O)C#N.Cl |
| InChi Key | PKGUOXDXRLGZBN-KUAPJGQRSA-N |
| InChi Code | InChI=1S/C10H12N4O4.ClH/c11-3-5-8(16)6(4-15)18-9(5)14-2-1-7(12)13-10(14)17;/h1-2,5-6,8-9,15-16H,4H2,(H2,12,13,17);1H/t5-,6+,8-,9+;/m0./s1 |
| Chemical Name | (2R,3S,4S,5R)-2-(4-amino-2-oxopyrimidin-1-yl)-4-hydroxy-5-(hydroxymethyl)oxolane-3-carbonitrile;hydrochloride |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment, avoid exposure to moisture. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | DNA (incorporation induces strand breaks) |
| ln Vitro |
The mechanism of action of CNDAC is distinct; upon entering DNA, it causes single-strand breaks (SSBs), which are subsequently transformed into double-strand breaks (DSBs) during the second S phase of cell division [1]. CNDAC (0-100 μM; 3 days) prevents HL-60 and THP-1 cells from swelling [2]. A number of cells, including Rad51D and XRCC3, are susceptible to CNDAC (0–1 μM; 24 h) [1]. After a delayed S phase, CNDAC (6 μM; 48 h) causes cell cycle arrest in HCT116 cells during the G2 phase [3]. -10 μM; 3–6 days) to cause HL-60 and THP-1 cells to become disinfected [2]. In clonogenic survival assays using Chinese hamster ovary (CHO) cell lines, CNDAC demonstrated potent cytotoxic activity. The IC₅₀ value for CNDAC in wild-type AA8 cells was 0.48 µM. Cells deficient in the homologous recombination (HR) repair pathway were significantly more sensitive to CNDAC. Specifically, Rad51D-deficient cells (51D1) showed an IC₅₀ of 0.006 µM, representing approximately 50-fold sensitization compared to Rad51D-complemented cells (51D1.3, IC₅₀ = 0.32 µM). XRCC3-deficient cells (irs1SF) exhibited even greater sensitivity, with an IC₅₀ of 0.0053 µM, which is about 89-fold more sensitive than wild-type AA8 cells (IC₅₀ = 0.48 µM). This stark contrast highlights that the cytotoxicity of CNDAC is highly dependent on functional HR for repair. In contrast, cells deficient in Rad51D or XRCC3 were not sensitized to cytarabine or gemcitabine (sensitization factor ~1). [1] The anti-proliferative and cytotoxic effects of CNDAC were evaluated against acute myeloid leukemia (AML) cell lines (HL-60 and THP-1) and primary AML cells from bone marrow (BM) and peripheral blood (PB). In the Alamar Blue assay, the IC₅₀ values for CNDAC against HL-60 and THP-1 cells were determined (Figure 1 and Table 2). For HL-60 cells plated at high density (0.5 × 10⁶ cells/mL), the IC₅₀ was 5.356 µM on day 3 and decreased to <0.5 µM by day 6. For HL-60 cells plated at low density (0.05 × 10⁶ cells/mL), the IC₅₀ was consistently <0.5 µM from day 3 to day 6. For THP-1 cells (which are less sensitive to Ara-C), the IC₅₀ for CNDAC was 0.929 µM (low density, day 3) and >10 µM (high density, day 3), decreasing to 1.104 µM and 2.095 µM respectively by day 6. [2] Using trypan blue exclusion assays, CNDAC induced significant cell death in both AML cell lines at concentrations ranging from 0.5 µM to 10 µM. In HL-60 cells (Ara-C sensitive), CNDAC was more effective than Ara-C at equivalent concentrations, especially at lower cell seeding densities. In THP-1 cells (Ara-C resistant), CNDAC showed significantly higher cell death than Ara-C at doses >2 µM. CNDAC demonstrated a delayed effect in THP-1 cells, with more robust cytotoxicity observed on days 4-6 compared to day 3. [2] Flow cytometry analysis (7-AAD/Annexin V staining) confirmed the induction of apoptosis by CNDAC in both cell lines. The distribution of early and late apoptotic events differed between HL-60 and THP-1 cells. [2] In primary AML mononuclear cells (MNCs) from 5 patients, CNDAC demonstrated potent cytotoxic activity. Cells were treated for 4 days, and survival was assessed up to 35 days post-treatment. Treatment with 10 µM (medium dose) CNDAC resulted in a significant and sustained reduction in cell survival for both PB and BM MNCs compared to untreated controls on days 4, 7, and 14. Even at a low dose (1 µM), CNDAC treatment led to significantly lower overall survival of PB and BM cells over the 35-day culture period compared to untreated cells and to cells treated with 1 µM Ara-C. Importantly, residual cells after CNDAC treatment did not expand in culture post-drug washout, unlike cells treated with Ara-C. [2] CNDAC showed potent growth inhibitory activity against murine leukemia L1210 cells with an IC₅₀ of 0.53 μM and against human oral epidermoid carcinoma KB cells with an IC₅₀ of 5.2 μM. It exhibited a broad spectrum of cytotoxicity against 15 human solid tumor cell lines, including lung adenocarcinoma (PC-8, PC-9), stomach adenocarcinoma (ST-KM, MKN-45), colon adenocarcinoma (Colo-320), breast adenocarcinoma (MCF-7), osteosarcoma (OST, MNNG/HOS), fibrosarcoma (HT-1080), and melanoma (A-375), with IC₅₀ values ranging from 0.53 μM to >360 μM. CNDAC was more cytotoxic than ara-C against several cell lines refractory to ara-C. N⁴-Acetyl-CNDAC (6b) was less potent than CNDAC, and other pyrimidine analogues (CNDAT, CNDAU) were inactive up to 100 μg/mL. The elimination product 10 (2′-C-cyano-2′,3′-didehydro-2′,3′-dideoxycytidine) was much less effective than CNDAC, indicating that the 3′-hydroxyl group is important for activity. CNDAA (adenine analogue) showed cytotoxicity against L1210 cells but was inactive against human solid tumor cell lines.[4] |
| ln Vivo |
In mice with active tumors, CNDAC (20 mg/kg; ip; daily for 10 days) demonstrated resistance [4]. CNDAC showed strong antitumor activity against M5076 mouse reticulum cell sarcoma when administered orally at 400 mg/kg/day on days 1, 4, 7, 10, 13, and 16, achieving 99% tumor volume inhibition on day 20 (T/C = 133%). At 200 mg/kg/day under the same schedule, it showed 93% tumor volume inhibition (T/C = 136%). Long-term survivors were observed in both dose groups. CNDAC was also highly effective against P388 mouse leukemia with T/C >600% and survival over 60 days in 5 out of 6 mice at 20 mg/kg/day intraperitoneally on days 1–10. It was effective against HT1080 human fibrosarcoma implanted in chick embryos or athymic mice, which is refractory to ara-C.[4] |
| Cell Assay |
Cell viability assay [1] Cell Types: Rad51D-deficient 51D1, Rad51D-complemented 51D1.3, wild-type AA8 and XRCC3-deficient irs1SF CHO cell Tested Concentrations: 0-1 μM Incubation Duration: 24 h Experimental Results: Inhibition of cell survival. The IC50 against Rad51D-deficient 51D1, Rad51D-complemented 51D1.3, wild-type AA8, and XRCC3-deficient irs1SF cell lines were 0.006, 0.32, 0.48, and 0.0053 μM, respectively. Cell proliferation assay [2] Cell Types: HL-60 and THP-1 Cell Tested Concentrations: 0-100 μM Incubation Duration: 3 days Experimental Results: Inhibitory effect on the proliferation of HL-60 and THP-1 cells, IC50 is 1.5832 μM and 0.84 μM, respectively. Apoptosis analysis[2] Cell Types: HL-60 and THP-1 Cell Tested Concentrations: 0, 0.5, 1, 2, 3, 4, 5 and 10 μM Incubation Duration: 3, 4, 5 and 6 days Experimental Results: Induction Apoptosis in both cells. Cell cycle analysis [3] Cell Types: HCT116 Tested Concentrations: 6 μM Incubation Duration: 48 hrs (hours) Experimental Results: 36% and 36% of cells were arrested in late S and G2/M phases respectively. Clonogenic survival assays were performed to evaluate the cytotoxicity of CNDAC and its dependence on DNA repair pathways. Exponentially growing CHO cells were seeded in 6-well plates and allowed to attach overnight. The following day, cells were exposed to a range of concentrations of CNDAC for 24 hours. After the treatment period, the drug-containing medium was removed, cells were washed, and fresh drug-free medium was added. Cells were then incubated for an additional 4 to 6 days to allow colony formation. Subsequently, colonies were fixed and stained with a crystal violet solution in ethanol. The stained plates were scanned, and colonies were counted electronically using a colony counting system. Survival curves were plotted, and IC₅₀ values were calculated from these curves. [1] Alamar Blue Assay for IC₅₀ Determination: HL-60 and THP-1 cells were plated in 96-well flat-bottom plates at 5 × 10³ cells/well and incubated for 24 hours. Cells were then treated with a range of concentrations of CNDAC (0.005 to 100 µM) in triplicate for 72 hours. After treatment, Alamar Blue reagent was added to each well at a final concentration of 10%. Plates were returned to the incubator for 8 hours. Absorbance was measured at 570 nm and 600 nm using a plate reader. The percentage reduction of Alamar Blue was calculated, and from this, the percentage inhibition of cell proliferation was determined. Non-linear regression standard curves were generated to calculate IC₅₀ values. [2] Cell Viability and Proliferation Assay (Trypan Blue Exclusion): Cell lines (HL-60, THP-1) were seeded in 48-well plates at two densities (0.05 × 10⁶ cells/mL and 0.5 × 10⁶ cells/mL). Cells were treated with CNDAC at concentrations ranging from 0.5 µM to 10 µM for up to 6 days. At designated time points (3, 4, 5, 6 days), cells were collected, mixed with trypan blue dye, and counted using a hemocytometer. The percentage of dead cells was calculated from raw counts. [2] Primary AML PB MNCs were treated with CNDAC (1, 10, 100 µM) in suspension for 4 days. Primary AML BM MNCs were treated with the same concentrations of CNDAC but in a co-culture system with irradiated M2-10B4 mouse stromal cells for 4 days. After treatment, cells were washed, re-plated onto fresh stromal layers, and cultured for an additional 31 days (total 35-day observation). Cell survival was assessed by trypan blue exclusion at days 4 (end of treatment), 7, 14, and 35. Survival percentage was calculated relative to the initial number of cells plated. [2] Apoptosis Assay (Flow Cytometry): Cell lines treated with CNDAC were collected, washed with cold PBS, and resuspended in binding buffer. Cells were then stained with Annexin V and 7-AAD according to the manufacturer's instructions, incubated in the dark for 15 minutes at room temperature, washed, and analyzed by flow cytometry within 1 hour. Cells were categorized as live (Annexin V⁻ 7-AAD⁻), early apoptotic (Annexin V⁺ 7-AAD⁻), and late apoptotic/necrotic (Annexin V⁺ 7-AAD⁺). [2] Tumor cell growth inhibitory activity was assessed using a 72-hour incubation assay. Cells (2 × 10³ per well) were incubated with test compounds, followed by addition of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide). After 4 hours, formazan was dissolved in DMSO and absorbance at 540 nm was measured. Percent inhibition was calculated as [1 − (OD of sample well / OD of control well)] × 100. IC₅₀ was defined as the concentration causing 50% inhibition of cell growth.[4] Cytotoxicity against various human tumor cell lines was tested similarly.[4] |
| Animal Protocol |
Animal/Disease Models: CDF1 mouse, P388 tumor model [4] Doses: 20 mg/kg Route of Administration: intraperitoneal (ip) injection, daily for 10 days Experimental Results: Greatly improved survival time and survival rate. M5076 reticulum cell sarcoma cells (10⁶) were implanted subcutaneously into the axillary region of female BD2F₁ mice. CNDAC was administered orally on days 1, 4, 7, 10, 13, and 16 at doses of 50, 100, 200, 300, and 400 mg/kg/day. Tumor volume was measured as 0.5 × length × width². Tumor growth inhibition and T/C (%) were calculated on day 20. Body weight changes were also recorded.[4] For P388 leukemia, CNDAC was administered intraperitoneally once daily on days 1–10 at 20 mg/kg/day.[4] |
| ADME/Pharmacokinetics |
The provided literature [2] does not describe ADME (Absorption, Distribution, Metabolism, Excretion) or pharmacokinetic properties (e.g., half-life, bioavailability) for CNDAC itself. It mentions that human pharmacokinetic studies have detected plasma concentrations of CNDAC upwards of 0.25 µM following administration of its prodrug, sapacitabine. [2] |
| References |
[1]. Sapacitabine, the prodrug of CNDAC, is a nucleoside analog with a unique action mechanism of inducing DNA strand breaks. Chin J Cancer. 2012 Aug;31(8):373-80. [2]. Bone Marrow and Peripheral Blood AML Cells Are Highly Sensitive to CNDAC, the Active Form of Sapacitabine. Adv Hematol. 2012;2012:727683. [3]. Antiproliferative effects of sapacitabine (CYC682), a novel 2'-deoxycytidine-derivative, in human cancer cells. Br J Cancer. 2007 Sep 3;97(5):628-36. [4]. Nucleosides and nucleotides. 122. 2'-C-cyano-2'-deoxy-1-beta-D-arabinofuranosylcytosine and its derivatives. A new class of nucleoside with a broad antitumor spectrum. J Med Chem. 1993 Dec 24;36(26):4183-9. |
| Additional Infomation |
Radgocitabine Hydrochloride is the hydrochloride salt form of radgocitabine, an analogue of the nucleoside deoxycytidine with potential antineoplastic activity. Upon administration, radgocitabine is incorporated into DNA and directly inhibits the activity of DNA polymerase, which may result in inhibition of DNA replication and cell cycle arrest in the S and G2/M phases, DNA fragmentation, and tumor cell apoptosis. CNDAC (2'-C-cyano-2'-deoxy-1-β-D-arabino-pentofuranosylcytosine) is the active metabolite of the orally bioavailable prodrug sapacitabine. Both are under clinical investigation for hematologic malignancies and solid tumors. [1] CNDAC has a unique mechanism of action among deoxycytidine analogs. After incorporation into DNA during replication, the cyanosugar moiety induces structural instability. This leads to a β-elimination rearrangement, resulting in a single-strand break (SSB) and the formation of a chain-terminating residue (CNddC) at the 3' end. These SSBs can be repaired by the transcription-coupled nucleotide excision repair (TC-NER) pathway. If unrepaired, when the cell enters a second S phase, the SSBs are converted into one-ended double-strand breaks (DSBs). These lethal DSBs are primarily repaired via the homologous recombination (HR) pathway. Deficiency in key HR components (e.g., Rad51D, XRCC3) profoundly sensitizes cells to CNDAC. [1] This distinct mechanism and repair dependency differentiate CNDAC from other deoxycytidine analogs like cytarabine and gemcitabine, which do not show this reliance on HR for repair. This suggests CNDAC/sapacitabine may have a different clinical application profile and could be effective in tumors with HR deficiencies or in overcoming resistance to other nucleoside analogs. [1] Clinical trials for sapacitabine (the prodrug) are ongoing in various cancers including acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), chronic lymphocytic leukemia (CLL), and non-small cell lung cancer (NSCLC). [1] CNDAC is the active metabolite of the oral prodrug sapacitabine. It is a deoxycytidine analog structurally related to cytarabine (Ara-C) and gemcitabine, distinguished by a cyano group replacing the 2' hydrogen on the sugar moiety. [2] CNDAC has a unique mechanism of action. After incorporation into DNA, the cyano group causes structural instability, leading to a β-elimination rearrangement. This generates a single-strand break (SSB) and a chain-terminating residue (CNddC) that lacks a 3'-OH group. These SSBs are poorly repaired (slow nucleotide excision repair process) and are converted into double-strand breaks (DSBs) during subsequent DNA replication, leading to cell death. [2] CNDAC is reported to be a poor substrate for cytidine deaminase (CDA), an enzyme that inactivates Ara-C, which may explain its activity in Ara-C resistant models like the THP-1 cell line. [2] The study suggests that CNDAC (delivered as sapacitabine) is more potent than Ara-C in vitro against AML cell lines and primary patient cells, including those from bone marrow niches protected by stroma. Its prolonged effect post-washout and activity at low doses highlight its potential clinical utility, particularly for elderly AML patients or those resistant to conventional Ara-C-based therapy. [2] Sapacitabine (the prodrug) is under clinical investigation (Phase III trials mentioned) for newly diagnosed AML in elderly patients. [2] CNDAC is a cytosine nucleoside analogue designed to have an electron-withdrawing cyano group at the 2′-β position. It is hypothesized to be phosphorylated to the 5′-triphosphate and incorporated into DNA, where the cyano group may promote β-elimination, leading to DNA strand breaks or abasic site formation, contributing to its antitumor mechanism.[4] It is chemically stable as a nucleoside but becomes reactive after incorporation into DNA.[4] CNDAC shows a different antitumor spectrum compared to ara-C and is effective against ara-C-resistant tumors.[4] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~125 mg/mL (~432.99 mM) |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.4639 mL | 17.3196 mL | 34.6392 mL | |
| 5 mM | 0.6928 mL | 3.4639 mL | 6.9278 mL | |
| 10 mM | 0.3464 mL | 1.7320 mL | 3.4639 mL |