Physicochemical Properties
| Molecular Formula | C28H25NO7S |
| Molecular Weight | 519.57 |
| Exact Mass | 519.135 |
| Elemental Analysis | C, 64.73; H, 4.85; N, 2.70; O, 21.56; S, 6.17 |
| CAS # | 249886-47-3 |
| PubChem CID | 9958278 |
| Appearance | Typically exists as solid at room temperature |
| LogP | 5.4 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 8 |
| Rotatable Bond Count | 10 |
| Heavy Atom Count | 37 |
| Complexity | 820 |
| Defined Atom Stereocenter Count | 0 |
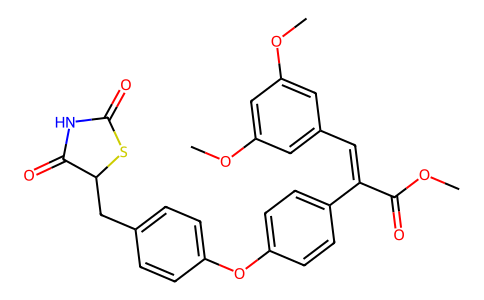
| SMILES | COC1=CC(=CC(=C1)/C=C(\C2=CC=C(C=C2)OC3=CC=C(C=C3)CC4C(=O)NC(=O)S4)/C(=O)OC)OC |
| InChi Key | IVAQJHSXBVHUQT-ZVHZXABRSA-N |
| InChi Code | InChI=1S/C28H25NO7S/c1-33-22-12-18(13-23(16-22)34-2)14-24(27(31)35-3)19-6-10-21(11-7-19)36-20-8-4-17(5-9-20)15-25-26(30)29-28(32)37-25/h4-14,16,25H,15H2,1-3H3,(H,29,30,32)/b24-14+ |
| Chemical Name | methyl (E)-3-(3,5-dimethoxyphenyl)-2-[4-[4-[(2,4-dioxo-1,3-thiazolidin-5-yl)methyl]phenoxy]phenyl]prop-2-enoate |
| Synonyms | CLX0921; THR-0921; CLX-0921; THR-0921; 4LXSR6QYJI; 606932-81-4; UNII-4LXSR6QYJI; 249886-47-3; 3-(3,5-Dimethoxyphenyl)-2-(4-(4-(2,4-dioxothiazolidin-5-ylmethyl)-phenoxy)-phenyl)-acrylic acid methyl ester; CLX-0921 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | PPARγ/peroxisome proliferator-activated receptor gamma (IC50 = 1.54 μM) |
| ln Vitro |
CLX-0921 is a new PPAR gamma agonist that is derived from a natural product. This thiazolidinedione (TZD) has a spectrum of activity that differs from commercially available TZDs. It is a weak activator of PPAR gamma (EC(50) of 0.284 micromol/L) compared to rosiglitazone (EC(50) 0.009 micromol/L). Despite this difference, the drug maintains potent glucose uptake activity in vitro and glucose-lowering activity in vivo that is equipotent to that of rosiglitazone. Moreover, CLX-0921 showed a 10-fold reduction in in vitro adipogenic potential compared to rosiglitazone. CLX-0921 also increases glycogen synthesis, an activity not typically associated with rosiglitazone or pioglitazone. Thus CLX-0921 appears to have a distinct spectrum of activity relative to other TZDs. [1] CLX-0921 stimulates glucose uptake In vitro [1] Differentiating 3T3-L1 adipocytes represent an insulin-sensitive cell-culture model for studying glucose uptake and is often used to characterize potential antidiabetic compounds. Although TZDs increase glucose uptake in these cells, both in the absence and presence of insulin,6, 13 the majority of this effect appears to be the result of non-insulin-mediated glucose disposal. As shown in Fig 2A, glucose uptake was increased to a maximum of 1.33 ± 0.02 (mean ± SE) fold over basal levels in response to increasing concentrations of CLX-0921 (0.01, 0.1, 1.0, and 10 μmol/L). We also examined the effect of CLX-0921 and rosiglitazone on insulin-stimulated glucose uptake in 3T3-L1 adipocytes (Fig 2B). There was no difference in dose-response curves of glucose uptake in response to insulin in the presence (5 μmol/L) or absence of the TZDs. Differences in the maximal responses can be accounted for by the increased amount of basal glucose uptake in the absence of insulin and either CLX-0921 or rosiglitazone, indicating an additive, not synergistic, effect on glucose uptake. These results suggest this enhancement of glucose uptake is mediated through a non-insulin-dependent mechanism, such as an increase in GLUT-1 transporters. |
| ln Vivo |
In vivo antihyperglycemic effect of CLX-0921[1] The antihyperglycemic activity of CLX-0921 was examined in several models of type 2 diabetes mellitus. Figure 3 summarizes the effect of CLX-0921 given as single daily oral doses of 50 mg/kg (96.2 μmol/kg) over 8 to 9 days. At the end of each study the drug led to marked decreases in blood glucose levels in ob/ob mice (59% v baseline), db/db mice (32% v baseline), and ZDF rats (50% v baseline). In ob/ob mice, CLX-0921 led to a 58% decrease in insulin levels after the 9-day dosing period compared to control (7.3 ± 1.1 v 17.6 ± 0.6 ng/mL, respectively, P < .001). Serum triglycerides and free fatty acids (FFA) were also lower in CLX-0921-treated ob/ob mice compared to controls by 64% (CLX-0921, 51.2 ± 6.7; control, 143 ± 7.4 mg/dL, P < .001) and 34% (CLX-0921, 1.75 ± 0.29; control, 2.64 ± 0.12 mg/dL, P < .05), respectively. Weight gain in treated and control animals was similar except for ZDF rats, where CLX-0921-treated animals gained 13% more weight than control animals. To examine the dose-response relationship between blood glucose and CLX-0921, ob/ob mice were treated once daily with vehichle or 3 different doses of CLX-0921 (3.1, 6.3, and 12.5 mg/kg body weight by oral gavage; n = 8 per group). As shown in Fig 4, there was a dose dependent decrease in blood glucose levels that was most apparent during the first week of treatment. Compared to the vehicle group, mean blood glucose levels decreased by 35%, 55%, and 47%, for the 3.1-, 6.3-, and 12.5-mg/kg dose groups, respectively, at day 19 (P ≤ .01, .001, and .001, respectively). In a subsequent study, the in vivo potency of CLX-0921 was compared to rosiglitazone in ob/ob mice (Fig 5). CLX-0921 and rosiglitazone treatment (both at 10 mg/kg/d; 19.2 and 28.0 μmol/kg/d for CLX-0921 and rosiglitazone, respectively) demonstrated similar antidiabetic potency over the 8-day treatment period. Weight gain was similar in vehicle and drug-treated groups. |
| Enzyme Assay |
PPARγ affinity by radioligand binding assay[1] Ligand binding assays were performed using human recombinant PPARγ as previously described Berger et al.11 Briefly, a GST-hPPARγ ligand binding domain chimeric cDNA construct was used to generate PPARγ in COS-1 cells. A radiolabled TZD ligand ([3H-AD-5075], KD ∼1 nmol/L) was incubated with transfected COS-1 cell lysates in the presence or absence of test compounds (CLX-0921 or rosiglitazone). Unbound ligand was removed with dextran/charcoal and supernatant fractions were measured in a scintillation counter. PPARγ cofactor protein association[1] A homogeneous time-resolved fluorescence assay (HTRF) was used to examine the interaction of liganded PPARγ and the coactivator protein CBP as described elsewhere.12 GST-PPARγ ligand binding domain (LBD), 2 nmol/L anti-GST-(Eu)K, 10 nmol/L biotin-CBP1-453, 20 nmol/L SA/XL665 were added to each well, followed by addition of test compound or vehicle (Me2SO) in individual wells. Plates were mixed by hand, covered and incubated overnight at 4°C. Fluorescence was measured on a Discovery instrument. This assay measures the ligand dependent interaction of PPARγ and CBP through energy transfer from (Eu)K to SA/XL665. |
| Cell Assay |
Glucose uptake[1] Basal glucose uptake was measured in differentiated 3T3-L1 adipocytes following the protocol of Tafuri6 with modifications. Briefly, 3T3-L1 fibroblasts, obtained from ATCC, were differentiated to adipocytes by treating cells with porcine insulin (1 μg/mL for 4 days), dexamethasone (0.25 μmol/L for first 2 days), and isobutyl methyl xanthine (IBMX, 0.5 mmol/L for first 2 days) following the protocol of Frost and Lane.7 Differentiated adipocytes were incubated in Dulbecco’s modified Eagle medium (DMEM) containing 10 % fetal bovine serum with various concentrations of CLX-0921 or vehicle (0.1% dimethyl sulfoxide [DMSO]) for 48 hours in 24-well plates, in triplicate. Cells were washed with phosphate-buffered saline ([PBS], 150 mmol/L NaCl, 1 mmol/L KH2PO4, 3 mmol/L Na2HPO4; pH 7.4) and incubated in glucose-free DMEM for 2 hours at 37°C. The cells were washed 3 times with Krebs Ringer phosphate buffer (KRP). Glucose uptake was initiated by addition of 0.25 μCi 2-14C(U)-deoxy-d-glucose (300 μCi/mmol) per well and the cells incubated for 10 minutes at room temperature in the presence of 0.1 mmol cold 2-deoxy-d-glucose. Finally, the cells were washed 3 times with ice-cold PBS containing 10 mmol/L cold glucose, lysed with 0.5 % sodium dodecyl sulfate (SDS), and counted in a scintillation counter. Glycogen synthesis[1] Glycogen synthesis was measured as net conversion of 14C-d-glucose to cellular glycogen in HepG2 cells as described by Ciaraldi et al.8 Briefly, HepG2 cells (ATCC) in 6-well plates were treated with CLX-0921 or other compounds for 48 hours. They were washed with 10 mmol/L HEPES buffer (150 mmol/L NaCl, 5 mmol/L KCl, 1.2 mmol/L MgSO4, 1.2 mmol/L CaCl2, 2.5 mmol/L CaCl2, 10 mmol/L HEPES; pH 7.4) containing 1% bovine serum albumin (BSA). Cells were incubated in the same buffer for 30 minutes prior to addition of 0.2 μCi/well of 14C-d-glucose (5 mmol/L; final concentration, 10 μCi/mmol). After incubation for 2 hours at 37°C, the cells were washed with ice-cold PBS and solubilized with 1 mol/L KOH at 55°C. Converted glycogen was precipitated by ethanol after addition of 10 mmol/L carrier glycogen. The pellet was washed and resuspended in water, and an aliquot was counted in a scintillation counter. Total protein was assayed and results were reported as cpm/mg of protein. Adipogenesis[1] Adipogenesis in 3T3-L1 fibroblasts was performed as described by Wu et al.9 After 2 days of growth in 6-well plates, cells were treated either with vehicle (0.1% DMSO) or with compounds for 14 days. Fresh medium with compounds or vehicle was replenished every 48 hours. Cells were washed with PBS twice and fixed in 10% formalin (Sigma) in PBS. After washing in PBS, cells were stained with freshly diluted Oil Red O in isopropanol for 1 hour at room temperature. The cells were washed 5 times with PBS and visualized under an Olympus BH2 microscope. Quantitative accumulation of triglyceride was also measured under similar experimental conditions, except in this case cells were plated in 100-mm tissue culture dishes and culture continued for 10 days. Triglyceride was extracted with methanol:chloroform (2:1) mixture. To monitor the efficiency of recovery, 3H-cholesterol oleate (50,000 cpm/well) was added in each tube as tracer before extraction following the protocol of Brown et al.10 Extracted triglyceride was measured by a colorimetric assay according to manufacturer’s instructions. Transfection and transactivation assays[1] Human PPARγ2 expression vector was constructed by inserting the PPARγ2 cDNA coding region into the pcDNA3.1+ vector. The PPRE-luciferase reporter gene was the kind gift of Dr Kenneth Feingold. The control vector, pRL-SV40, containing the Renilla luciferase cDNA was purchased from Promega. About 2.7 × 104 HEK293 human embryonal kidney cells (ATCC) were plated into a 35-mm tissue culture dish and maintained in Eagle modified essential medium (EMEM, ATCC) containing 10% heat-inactivated horse serum for 24 hours. Expression, reporter (100 ng/dish), and control (2.5 ng/dish) vectors were transfected using LIPOFECTAMIN PLUS Reagent according to manufacturer’s recommendation. At 24 hours after transfection, cells were treated with vehicle (0.001% DMSO in medium) or compounds at the indicated concentration and incubated for 24 hours. Each treatment was conducted in triplicate. Each culture dish was assayed for firefly luciferase activity normalized by Renillla luciferase activity to account for differences in transfection efficiency. Luciferase activity was measured using the Dual-luciferase Reporter Assay System and a Sirius luminometer. |
| Animal Protocol |
In vivo studies[1] Animals were housed at 22°C and 50% relative humidity, with a 12-hour light and dark cycle, and received a regular rodent diet ad libitum with free access to water. Male C57BL/KsJ-db/db and C57BL/6J-ob/ob mice were obtained from Jackson Laboratories at age 5 weeks. Seven-week-old animals (6 animals per group) were dosed with CLX-0921, rosiglitazone (purified from commercially available tablets), or vehicle (0.5% carboxymethyl cellulose in water) orally once daily by gavage. Blood glucose measurements were made with a One Touch Glucose Meter and/or a glucose oxidase assay prior to administering the next dose and in the fed state. Body weights were monitored throughout the study. Eight-week-old male Zucker diabetic fatty (ZDF-fa/fa) rats were kept on a 6.5% fat Formulab Diet 5008 for 2 weeks prior to dosing as described above. |
| References | [1]. A novel peroxisome proliferator-activated gamma (PPAR gamma) agonist, CLX-0921, has potent antihyperglycemic activity with low adipogenic potential. Metabolism. 2003 Aug;52(8):1012-8. |
| Additional Infomation |
CLX-0921 is investigated for use/treatment in diabetes mellitus type 2. CLX-0921 is a solid. CLX-0921 has a spectrum of activity that differs from commercially available thiazolidinediones. This substance targets the protein peroxisome proliferator-activated receptor gamma. It is a pharmacologically active antihyperglycemic agent that acts by increasing peripheral tissue sensitivity to insulin. Drug Indication Investigated for use/treatment in diabetes mellitus type 2. Mechanism of Action CLX-0921 has a spectrum of activity that differs from commercially available TZDs. It also increases glycogen synthesis, an activity not typically associated with rosiglitazone or pioglitazone. Thus it appears to have a distinct spectrum of activity relative to other TZDs. The affinity (Ki) of PPARγ for CLX-0921 was 6.5-fold less than its affinity for rosiglitazone (Table 1). The transactivation potency was as much as 30-fold less than that of rosiglitazone. In general, there is a relatively strong correlation between PPARγ affinity and glucose-lowering activity11; however, recent data indicate that this relationship may not be true for all ligands of this receptor. Recently a non-TZD PPARγ activator, FMOC-l-Leucine, has been shown to have a similar profile to CLX-0921. FMOC-l-Leucine has approximately 400-fold less affinity for PPARγ, is only weakly adipogenic, but has potent in vivo antihyperglycemic activity.22 Differences in the in vivo metabolism may explain part of the apparent discrepancy between PPARγ affinity and in vivo antidiabetic potency for CLX-0921 and other ligands. Studies by Reginato et al, Mukherjee et al,and Rocchi et al indicate that ligand-mediated recruitment of the coactivator SRC-1 to PPARγ is an important determinant for differential activities of ligands. At present, we can only speculate on the way in which CLX-0921 influences coactivator recruitment to the PPARγ-RXR complex. It may be that CLX-0921 is less conducive for SRC-1 or PGC-123 recruitment and, as a result, transcriptional activation. A recent study14 reported that in vitro glucose uptake into adipocytes is partially independent of PPARγ. Whether non-PPARγ-mediated activities or coactivator recruitment explains the unique properties of CLX-0921 will require further investigation. Presently, the mechanism by which PPARγ ligands, including TZDs, produce their antihyperglycemic effects is not known. The prevailing wisdom suggests that the glucose-lowering effect of these drugs is mediated through the PPARγ receptor, which enhances insulin sensitivity. Recent data suggest that the relationship between PPARγ, its ligands, and insulin sensitivity is more complex. For example, heterozygous PPARγ null mice actually demonstrate increased insulin sensitivity, and the insulin-sensitizing effect of synthetic ligands may result from a balance between transcriptional activation and repression. Abrogating endogenous PPARγ does not result in the elimination of TZD activity, which further highlights the complexity of the mechanism of action of these drugs. At present, a non-receptor-mediated mechanism of action for CLX-0921 and other PPARγ agonists cannot be excluded. However, we have been unable to demonstrate any insulin receptor-mediated activity of CLX-0921, which was expected based on the compound’s structural derivation from CLX-0901. Our data indicate that CLX-0921, in contrast to rosiglitazone, increases glycogen synthesis in liver cells, possibly providing an added mechanism for lowering glucose levels in diabetic animals. Recent data suggest that some non-TZD PPARγ agonists may upregulate genes involved in glycogen synthesis. It is becoming apparent that ligands for this receptor will have a spectrum of affinities, transcriptional activities, and in vivo pharmacodynamic profiles. Therefore, there is substantial clinical value in generating compounds with selective PPARγ modulating activities. The acronym SPPRM, as coined by John Auwerx’s group for “selective PPAR modulator,” may best describe these molecules. SPPRMs may ultimately prove to have both specific and tailored activities, including the potential avoidance of weight gain associated with currently marketed TZDs. Such agents would have the potential to be of great benefit in treating patients with type 2 diabetes.[1] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.9247 mL | 9.6233 mL | 19.2467 mL | |
| 5 mM | 0.3849 mL | 1.9247 mL | 3.8493 mL | |
| 10 mM | 0.1925 mL | 0.9623 mL | 1.9247 mL |