CDC801 is a novel, potent and orally bioavailable inhibitor of phosphodiesterase 4 (PDE4) and tumor necrosis factor-α (TNF-α) with IC50 of 1.1 μM and 2.5 μM, respectively.
Physicochemical Properties
| Molecular Formula | C23H24N2O5 |
| Molecular Weight | 408.447066307068 |
| Exact Mass | 408.168 |
| CAS # | 192819-27-5 |
| PubChem CID | 9844338 |
| Appearance | White to off-white solid powder |
| LogP | 2.5 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 5 |
| Rotatable Bond Count | 7 |
| Heavy Atom Count | 30 |
| Complexity | 638 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | DDYUBCCTNHWSQM-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C23H24N2O5/c1-29-19-11-10-14(12-20(19)30-15-6-2-3-7-15)18(13-21(24)26)25-22(27)16-8-4-5-9-17(16)23(25)28/h4-5,8-12,15,18H,2-3,6-7,13H2,1H3,(H2,24,26) |
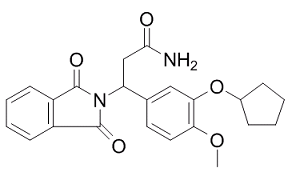
| Chemical Name | 3-(3-(cyclopentyloxy)-4-methoxyphenyl)-3-(1,3-dioxoisoindolin-2-yl)propanamide |
| Synonyms | CDC-801 CDC 801 CDC801 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
1. Phosphodiesterase 4 (PDE4, pan-isoform inhibitor, Ki = 11 nM for PDE4B; IC50 = 26 nM for PDE4D) [1] 2. Tumor necrosis factor-alpha (TNF-α, inhibitor of its secretion in immune cells, EC50 = 300 nM for LPS-induced TNF-α suppression in human monocytes) [1] |
| ln Vitro |
More specific information can be obtained in reference 1 for compound 6b (PMID: 19256507) [1]. 1. PDE4 enzyme inhibitory activity: CDC801 exhibited potent and selective inhibitory activity against PDE4 isoforms (PDE4A/B/C/D) with no significant inhibition of other PDE families (PDE1-PDE3, PDE5-PDE10) at concentrations up to 10 μM. The compound showed a Ki of 11 nM for PDE4B (the most pharmacologically relevant isoform) and an IC50 of 26 nM for PDE4D, with competitive binding to the enzyme’s cAMP-binding pocket [1] 2. Pro-inflammatory cytokine suppression: In LPS-stimulated human peripheral blood monocytes (PBMCs), CDC801 dose-dependently inhibited the secretion of TNF-α, with an EC50 of 300 nM for 50% reduction of cytokine levels. At 1 μM, it also suppressed the production of IL-12 (by 45%) and IL-23 (by 38%) without affecting the secretion of anti-inflammatory IL-10 (change < 10%) [1] 3. Intracellular cAMP elevation: In human THP-1 monocytic cells, CDC801 (100 nM-1 μM) increased intracellular cAMP levels in a dose-dependent manner; at 500 nM, cAMP concentrations were elevated by 2.8-fold relative to the control, confirming its PDE4-mediated mechanism of action [1] |
| ln Vivo |
1. Anti-inflammatory activity in rodent models: In LPS-induced murine endotoxemia models, oral administration of CDC801 (10 mg/kg and 30 mg/kg) reduced serum TNF-α levels by 52% and 78% respectively at 4 h post-LPS challenge, with a dose-dependent attenuation of systemic inflammatory signs (lethargy, hypothermia). In rat adjuvant-induced arthritis models, daily oral administration of CDC801 (30 mg/kg) for 14 days reduced paw edema by 65% and joint inflammation scores by 58% compared with the vehicle control [1] 2. Immunomodulatory effect in skin inflammation models: In murine oxazolone-induced contact dermatitis models, CDC801 (20 mg/kg, oral, once daily for 7 days) reduced ear swelling by 42% and dermal neutrophil infiltration (by 51%) via suppression of local TNF-α and IL-17 production [1] |
| Enzyme Assay |
1. Recombinant PDE4 enzyme activity assay: The assay was conducted in a buffer system containing purified recombinant PDE4 isoforms (PDE4B/PDE4D), cAMP substrate (radiolabeled or fluorescently tagged), and serial dilutions of CDC801 (0.1 nM-10 μM). The reaction was initiated by adding the enzyme and incubated at 30℃ for 30 min, then terminated by heating or adding a stop solution. The remaining cAMP was quantified using either scintillation counting (for radiolabeled substrate) or fluorescence resonance energy transfer (FRET)-based detection. Enzyme activity was calculated relative to the vehicle control, and Ki/IC50 values were derived from dose-response curve fitting [1] 2. PDE family selectivity assay: A panel of recombinant PDE isoforms (PDE1-PDE10) was incubated with CDC801 at concentrations up to 10 μM in the same reaction system as the PDE4 assay. Residual enzyme activity was measured to evaluate the compound’s selectivity; activity reduction < 10% was defined as no significant inhibition [1] |
| Cell Assay |
1. PBMC cytokine secretion assay: Human peripheral blood monocytes were isolated and seeded in 24-well plates (1×10⁶ cells/well) and pre-incubated with CDC801 (10 nM-10 μM) for 1 h. The cells were then stimulated with LPS (1 μg/mL) for 24 h under normoxic conditions. The culture supernatant was collected, and the concentrations of TNF-α, IL-12, IL-23, and IL-10 were measured using a sandwich enzyme-linked immunosorbent assay (ELISA). The absorbance at 450 nm was recorded, and cytokine concentrations were calculated via standard curves to assess the compound’s anti-inflammatory effect [1] 2. Intracellular cAMP measurement assay: THP-1 monocytic cells were seeded in 96-well plates (5×10⁴ cells/well) and treated with CDC801 (100 nM-1 μM) for 2 h. A cAMP detection reagent was added to the cells, and the plate was incubated at room temperature for 1 h in the dark. Fluorescence intensity was measured using a microplate reader with excitation/emission wavelengths specific to the detection reagent, and cAMP concentrations were calculated using a standard curve [1] |
| Animal Protocol |
1. LPS-induced endotoxemia mouse model and administration: Male C57BL/6 mice (6-8 weeks old, 20-25 g) were randomly divided into 3 groups (vehicle control, 10 mg/kg CDC801, 30 mg/kg CDC801), with 8 mice per group. CDC801 was dissolved in a mixture of DMSO and 0.5% methylcellulose (final DMSO concentration < 0.5%) to prepare the oral suspension. The compound was administered via oral gavage at a volume of 10 μL/g body weight, 1 h prior to intraperitoneal LPS injection (5 mg/kg). The vehicle group received the same volume of DMSO-methylcellulose mixture without CDC801. Serum was collected at 4 h post-LPS challenge for TNF-α quantification [1] 2. Adjuvant-induced arthritis rat model and administration: Male Lewis rats (10-12 weeks old, 200-250 g) were injected with complete Freund’s adjuvant (CFA) into the right hind paw to induce arthritis. Rats were randomly grouped (vehicle, 10 mg/kg CDC801, 30 mg/kg CDC801) 3 days post-CFA injection, with 6 rats per group. CDC801 was formulated as an oral suspension (same solvent as the mouse model) and administered daily via oral gavage for 14 days. Paw volume was measured using a plethysmometer every 3 days, and joint inflammation scores were evaluated using a 4-point scale at the end of the administration period [1] |
| ADME/Pharmacokinetics |
1. Oral bioavailability and absorption: After a single oral administration of CDC801 (10 mg/kg) to rats, the oral bioavailability was determined to be 73%, with a peak plasma concentration (Cmax) of 1.2 μM achieved at 1.5 h post-dose (Tmax = 1.5 h) [1] 2. Plasma half-life and distribution: The terminal plasma half-life (t1/2) of CDC801 in rats was 6.8 h, with a volume of distribution (Vd) of 0.8 L/kg, indicating moderate tissue distribution. The compound showed good penetration into inflamed joint tissues, with joint/plasma concentration ratio of 1.2 at 4 h post-dose [1] 3. Metabolic stability: CDC801 exhibited good metabolic stability in human liver microsomes, with an intrinsic clearance rate of 12 mL/min/kg and a half-life of 45 min. The major metabolic pathway was confirmed to be sulfonamide hydrolysis and aromatic ring hydroxylation via LC-MS/MS analysis [1] |
| Toxicity/Toxicokinetics |
1. Plasma protein binding: The plasma protein binding rate of CDC801 in human and rat plasma was measured via ultrafiltration, with binding rates of 88% (human) and 85% (rat), indicating high but reversible protein binding [1] 2. Acute in vivo toxicity: After a single oral dose of CDC801 (up to 200 mg/kg) in mice, no significant mortality or gross organ damage was observed within 7 days. At doses > 100 mg/kg, transient decreases in locomotor activity were noted but resolved within 24 h, with no long-term toxicity to liver or kidney (serum ALT/AST and creatinine levels within normal ranges) [1] 3. In vitro cytotoxicity: CDC801 showed no significant cytotoxicity to human PBMCs or THP-1 cells at concentrations up to 10 μM (cell viability > 90% after 72 h incubation) [1] |
| References | [1]. Man HW, et al. Discovery of (S)-N-[2-[1-(3-ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl] acetamide (apremilast), a potent and orally active phosphodiesterase 4and tumor necrosis factor-alpha inhibitor. J Med Chem. |
| Additional Infomation |
CDC-801 is under investigation in clinical trial NCT00006097 (Chemotherapy in Treating Patients With Chronic Lymphocytic Leukemia). Selective Cytokine Inhibitory Drug CC-1088 is an analog of thalidomide with potential antineoplastic activity that belongs to the functional class of agents called selective cytokine inhibitory drugs (SelCIDs). SelCIDs inhibit phosphodiesterase-4 (PDE 4), an enzyme involved in tumor necrosis factor alpha (TNF alpha) production. CC-1088 inhibits production of the cytokines vascular endothelial growth factor (VEGF) (a pro-angiogenic factor) and interleukin-6 (IL-6). (NCI04) 1. CDC801 (subsequently named apremilast) is a small-molecule, orally active PDE4 inhibitor with additional TNF-α-suppressive activity, developed as an anti-inflammatory and immunomodulatory agent for autoimmune and inflammatory diseases [1] 2. The mechanism of action of CDC801 involves selective inhibition of PDE4, which increases intracellular cAMP levels in immune cells; elevated cAMP activates protein kinase A (PKA) and exchange protein activated by cAMP (EPAC), leading to the suppression of pro-inflammatory transcription factors (NF-κB, AP-1) and subsequent reduction of TNF-α, IL-12, and IL-23 secretion [1] 3. CDC801 was advanced to clinical development due to its balanced potency, oral bioavailability, and favorable safety profile, with indications including psoriasis, psoriatic arthritis, and Behçet’s disease (later approved under the trade name Otezla) [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~16.67 mg/mL (~40.81 mM) |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4483 mL | 12.2414 mL | 24.4828 mL | |
| 5 mM | 0.4897 mL | 2.4483 mL | 4.8966 mL | |
| 10 mM | 0.2448 mL | 1.2241 mL | 2.4483 mL |