BX912 (BX-912) is a novel, potent and specific inhibitor of PDK-1 (phosphoinositide-dependent kinase 1) inhibitor with potential antitumor activity. In cell-free assays, it exhibits selectivity for PDK1 over PKA and PKC of 9 and 105 folds, respectively, and inhibits PDK-1 with an IC50 of 12 nM. By specifically targeting the PDK1/Akt signaling pathway, it demonstrated strong antiproliferative activity in vitro. This pathway is a promising target for anticancer medications because it is essential for the growth, survival, and angiogenesis of cancer cells.
Physicochemical Properties
| Molecular Formula | C20H23BRN8O |
| Molecular Weight | 471.35 |
| Exact Mass | 470.117 |
| Elemental Analysis | C, 50.96; H, 4.92; Br, 16.95; N, 23.77; O, 3.39 |
| CAS # | 702674-56-4 |
| Related CAS # | 702674-56-4 |
| PubChem CID | 11754511 |
| Appearance | White solid powder |
| Density | 1.6±0.1 g/cm3 |
| Index of Refraction | 1.750 |
| LogP | 1.38 |
| Hydrogen Bond Donor Count | 4 |
| Hydrogen Bond Acceptor Count | 6 |
| Rotatable Bond Count | 7 |
| Heavy Atom Count | 30 |
| Complexity | 549 |
| Defined Atom Stereocenter Count | 0 |
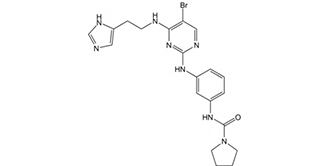
| SMILES | BrC1=C(NCCC2=CNC=N2)N=C(NC3=CC(NC(N4CCCC4)=O)=CC=C3)N=C1 |
| InChi Key | DMMILYKXNCVKOJ-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C20H23BrN8O/c21-17-12-24-19(28-18(17)23-7-6-16-11-22-13-25-16)26-14-4-3-5-15(10-14)27-20(30)29-8-1-2-9-29/h3-5,10-13H,1-2,6-9H2,(H,22,25)(H,27,30)(H2,23,24,26,28) |
| Chemical Name | N-(3-((4-((2-(1H-imidazol-4-yl)ethyl)amino)-5-bromopyrimidin-2-yl)amino)phenyl)pyrrolidine-1-carboxamide |
| Synonyms | BX 912; BX912; BX912 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
PDK-1 (IC50 = 2 nM); PKA (IC50 = 110 nM); KDR (IC50 = 410 nM); CDK2/CyclinE (IC50 = 650 nM); Chk1 (IC50 = 830 nM) The target of BX-912 is 3-phosphoinositide-dependent kinase-1 (PDK1). For recombinant human PDK1, the half-maximal inhibitory concentration (IC50) of BX-912 is approximately 6 nM. BX-912 shows high selectivity for PDK1: it has no significant inhibitory activity against a panel of other kinases, including Akt1 (IC50 > 10 μM), PKCα (IC50 > 10 μM), PKA (IC50 > 10 μM), and ERK2 (IC50 > 10 μM), indicating minimal off-target effects [1] |
| ln Vitro |
BX912 prevents ChcK1, PKA, c-kit, and KDR with IC50 of 0.83, 0.11, 0.85, and 0.41 μM, resepectively. BX912 inhibits PDK1/Akt signaling in tumor cells and either induces apoptosis or suppresses the anchorage-dependent growth of several tumor cell lines, including PC-3 cells, in culture. The PDK1/Akt signaling pathway's role in cell survival is supported by the fact that a number of cancer cell lines (including the MDA-468 breast cancer cell line) with elevated Akt activity are >30-fold more sensitive to growth inhibition by PDK1 inhibitor BX912 in soft agar than on tissue culture plastic, which is crucial for unattached cells in particular. In a direct kinase assay format, BX912 effectively inhibits PDK1 enzyme activity, but it is unable to inhibit preactivated AKT2 activity (IC50 > 10 μM). BX-912 is therefore a PDK1 direct inhibitor. BX912 may bind to the ATP-binding pocket of PDK1 because it inhibits the enzyme's ability to use its substrate, ATP, in a competitive manner. The aminopyrimidine backbone of BX912 adopts a similar orientation in the active site of PDK1. BX912 promotes a pronounced increase in the population of MDA-468 cells with 4 N DNA content, indicative of a block at the G2/M phase of the cell cycle. BX912 also potently inhibits the growth of HCT-116 cells in soft agar, showing a 96% inhibitory effect at a dose of 1 μM. BX912 potently inhibits the growth of PC-3 cells in soft agar, displaying IC50 of 0.32 μM. [1] 1. PDK1 enzyme activity inhibition: In vitro assays using purified recombinant human PDK1 showed that BX-912 dose-dependently inhibited PDK1-mediated phosphorylation of its specific substrate peptide (PDKtide, sequence: H-His-Arg-Ser-Gly-Arg-Thr-Ser-Phe-Ser-Ala-Arg-Lys-OH). The inhibition curve fitted from data of 8 concentration gradients (0.1 nM to 100 nM) confirmed an IC50 of ~6 nM, consistent with target binding specificity [1] 2. Inhibition of PDK1 downstream signaling in cancer cells: Treatment of HCT116 colon cancer cells with BX-912 (0.5 μM, 2 μM, 10 μM) for 4 hours resulted in a concentration-dependent reduction in the phosphorylation level of Akt (a key downstream substrate of PDK1) at Thr308 (detected by Western blot). Specifically, 2 μM BX-912 inhibited ~70% of Akt Thr308 phosphorylation, while the total Akt protein level remained unchanged. Similar results were observed in HeLa cervical cancer cells: 1 μM BX-912 reduced Akt Thr308 phosphorylation by ~50% after 6 hours of treatment [1] 3. Antiproliferative activity against cancer cells: Using the MTT assay to evaluate cell viability, BX-912 exhibited dose-dependent antiproliferative effects on multiple human cancer cell lines. For HCT116 colon cancer cells, the IC50 for 72-hour proliferation inhibition was approximately 2.1 μM; for HeLa cervical cancer cells, the IC50 was ~3.3 μM; and for A549 lung adenocarcinoma cells, the IC50 was ~4.5 μM. In contrast, BX-912 had no significant effect on the viability of normal human foreskin fibroblasts (NHFF) even at concentrations up to 10 μM, suggesting preferential activity against cancer cells [1] |
| ln Vivo | NA |
| Enzyme Assay |
AKT2 is activated by PDK1 and PtdIns-3,4-P2 in a PDK1 and coupled assay format. For the coupled assay, the final assay mixture (60 μL) contained: 15 mM MOPS, pH 7.2, 1 mg/mL bovine serum albumin, 18 mM β-glycerol phosphate, 0.7 mM dithiothreitol, 3 mM EGTA, 10 mM MgOAc, 7.5 μM ATP, 0.2 μCi of [γ- 33P]ATP, 7.5 μM biotinylated peptide substrate (biotin-ARRRDGGGAQPFRPRAATF), 0.5 μL of PtdIns-3,4-P2-containing phospholipid vesicles, 60 pg of purified recombinant human PDK1, and 172 ng of purified recombinant human AKT2. The biotin-labeled peptide is captured from 10 L of the assay mixture on streptavidin-coated SPA beads after incubating for 2 hours at room temperature. Product formation is then measured by scintillation proximity in a Wallac MicroBeta counter. The amount of PDK1 and inactive AKT2 added, along with the time of incubation, determine the amount of product that forms. PDK1 is added at suboptimal levels so that the assay could sensitively detect inhibitors of AKT2 activation as well as direct inhibitor BX912 of PDK1 or AKT2. To measure PDK1 activity directly, the final assay mixture (60 μL) contained 50 mM Tris-HCl, pH 7.5, 0.1 mM EGTA, 0.1 mM EDTA, 0.1% β-mercaptoethanol, 1 mg/mL bovine serum albumin, 10 mM MgOAc, 10 μM ATP, 0.2 μCi of [γ-33P]ATP, 7.5 μM substrate peptide (H2N-ARRRGVTTKTFCGT), and 60 ng of purified recombinant human PDK1. After 4 hours at room temperature, 25 mM EDTA is added, and some of the reaction mixture is spotted on P81 phosphocellulose paper. Three times in 0.75% phosphoric acid and once in acetone are used to wash the filter paper. A phosphorimager is used to measure the labeled peptide that is bound to the filter after drying. 1. Reagent preparation: Purified recombinant human PDK1 (expressed in Sf9 insect cells and purified via affinity chromatography) was prepared first. A reaction buffer containing 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 1 mM dithiothreitol (DTT), and 0.1 mg/mL bovine serum albumin (BSA) was formulated. The specific PDK1 substrate peptide (PDKtide) was dissolved in the reaction buffer to a final concentration of 100 μM. [γ-32P]ATP (radioactive ATP) was added to the reaction system to a final concentration of 10 μM, with a specific activity of ~1000 cpm/pmol [1] 2. Assay setup: BX-912 was serially diluted in dimethyl sulfoxide (DMSO) to obtain 8 concentration gradients (0.1 nM, 0.3 nM, 1 nM, 3 nM, 10 nM, 30 nM, 100 nM, 300 nM). Each dilution was added to the reaction buffer, ensuring the final DMSO concentration was ≤ 1% (this concentration has no effect on PDK1 activity). The reaction was initiated by adding recombinant PDK1 (final concentration 2 nM) to the mixture of BX-912, PDKtide, and [γ-32P]ATP. A blank control (containing DMSO but no BX-912) and a positive control (a known non-selective kinase inhibitor) were included. Each reaction was performed in triplicate in 96-well plates, and the plates were incubated at 30°C for 30 minutes [1] 3. Detection and data analysis: After incubation, 20 μL of each reaction mixture was spotted onto phosphocellulose filter papers. The filters were washed three times with 1% phosphoric acid (5 minutes per wash) to remove unincorporated [γ-32P]ATP, then rinsed once with acetone and air-dried. The radioactivity on the filters was measured using a liquid scintillation counter. The percentage of PDK1 inhibition was calculated using the formula: [(Radioactivity of blank control - Radioactivity of sample) / Radioactivity of blank control] × 100%. The IC50 value was determined by fitting the inhibition percentages to a four-parameter logistic regression model [1] |
| Cell Assay |
Cells such as MDA-468, MDA-453 are seeded at a low density (1.5-3 × 103 cells/well, 0.1 mL/well, 96-well plates) and are incubated overnight. BX912 treatments are made by adding 10 μL/well of the compound in 1% dimethyl sulfoxide and growth medium (final concentration of dimethyl sulfoxide, 0.1%), followed by brief shaking. The metabolic dye WST-1 is added to treated cells after 72 hours of incubation to determine viability. The net signal is calculated by subtracting a no cell, or zero time cell, background from the WST-1 signal, which is read at 450 nm on a plate reader. 1. Antiproliferation assay (MTT method): - Cell seeding: Human cancer cell lines (HCT116, HeLa, A549) and normal NHFF cells were trypsinized and resuspended in complete medium (DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin). Cells were seeded into 96-well plates at a density of 5 × 10³ cells per well and incubated at 37°C with 5% CO₂ for 24 hours to allow attachment [1] - Drug treatment: BX-912 was diluted with complete medium to 8 concentration gradients (0.1 μM, 0.3 μM, 1 μM, 3 μM, 10 μM, 30 μM, 100 μM), with the final DMSO concentration ≤ 1%. The medium in each well was replaced with 100 μL of the diluted BX-912 solution, and a vehicle control (medium containing 1% DMSO) was set up. Each concentration was tested in triplicate, and the plates were incubated for 72 hours [1] - Viability detection: After incubation, 20 μL of MTT solution (5 mg/mL in PBS) was added to each well, and the plates were incubated for another 4 hours. The supernatant was carefully aspirated, and 150 μL of DMSO was added to each well to dissolve the formazan crystals. The absorbance at 570 nm was measured using a microplate reader. The cell viability percentage was calculated as (Absorbance of sample / Absorbance of vehicle control) × 100%, and the IC50 for antiproliferation was obtained by fitting the viability data to a logistic model [1] 2. Western blot assay for PDK1 downstream signaling: - Cell preparation and treatment: HCT116 or HeLa cells were seeded into 6-well plates at a density of 2 × 10⁵ cells per well and cultured at 37°C with 5% CO₂ until reaching 70-80% confluence. The medium was replaced with fresh medium containing BX-912 at concentrations of 0.5 μM, 2 μM, and 10 μM (vehicle control: medium with 1% DMSO). The cells were incubated for 4-6 hours [1] - Protein extraction and electrophoresis: Cells were washed twice with ice-cold PBS, and 200 μL of RIPA lysis buffer (supplemented with protease and phosphatase inhibitors) was added to each well. The cells were scraped off on ice, and the lysates were centrifuged at 12,000 × g for 15 minutes at 4°C to collect the supernatant (total protein). The protein concentration was determined using a BCA assay. Equal amounts of protein (30 μg per lane) were separated by 10% SDS-PAGE and transferred to a PVDF membrane [1] - Immunodetection: The PVDF membrane was blocked with 5% non-fat milk in TBST for 1 hour at room temperature, then incubated overnight at 4°C with primary antibodies against phosphorylated Akt (Thr308), total Akt, and β-actin (loading control). After washing three times with TBST, the membrane was incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 hour at room temperature. The membrane was washed again, and protein bands were visualized using an enhanced chemiluminescence (ECL) reagent. The band intensity was quantified using image analysis software, and the ratio of phosphorylated Akt to total Akt was calculated to evaluate the inhibition of PDK1 downstream signaling by BX-912 [1] |
| Animal Protocol | NA; |
| References |
[1]. Novel small molecule inhibitors of 3-phosphoinositide-dependent kinase-1. J Biol Chem. 2005 May 20;280(20):19867-74. |
| Additional Infomation |
N-[3-[[5-bromo-4-[2-(1H-imidazol-5-yl)ethylamino]-2-pyrimidinyl]amino]phenyl]-1-pyrrolidinecarboxamide is a member of ureas. 1. BX-912 is one of the earliest reported selective small-molecule inhibitors of PDK1, designed to target the ATP-binding pocket of PDK1. It exerts its inhibitory effect by competing with ATP for binding to the catalytic domain of PDK1, thereby blocking PDK1’s ability to phosphorylate and activate downstream kinases (e.g., Akt, PKC) in the PI3K-Akt signaling pathway [1] 2. In cancer research, BX-912 serves as a valuable tool compound for dissecting the biological functions of PDK1 and its role in the progression of cancers with dysregulated PI3K-Akt signaling (e.g., colon cancer, cervical cancer). The compound’s high selectivity for PDK1 and preferential antiproliferative activity against cancer cells (compared to normal cells) support its potential as a lead compound for developing PDK1-targeted anticancer agents, though it has not advanced to clinical development [1] 3. The development of BX-912 provided a critical experimental tool to validate PDK1 as a therapeutic target. Studies using BX-912 demonstrated that inhibiting PDK1 can effectively suppress the activation of the PI3K-Akt pathway in cancer cells, leading to reduced cell proliferation—confirming PDK1’s role as a key mediator of cancer cell survival and growth [1] |
Solubility Data
| Solubility (In Vitro) |
DMSO: ~94 mg/mL (~199.4 mM) Water: <1 mg/mL Ethanol: N/A |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.75 mg/mL (5.83 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 27.5 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.75 mg/mL (5.83 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 27.5 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.75 mg/mL (5.83 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 27.5 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 30% PEG400+0.5% Tween80+5% propylene glycol: 30mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1216 mL | 10.6078 mL | 21.2157 mL | |
| 5 mM | 0.4243 mL | 2.1216 mL | 4.2431 mL | |
| 10 mM | 0.2122 mL | 1.0608 mL | 2.1216 mL |