BX795 is a novel, potent and selective/specific PDK-1 (3-phosphoinositide-dependent kinase-1) inhibitor with potential antitumor activity. In cell-free assays, it exhibits 140- and 1600-fold more selectivity for PDK1 over PKA and PKC, respectively, and inhibits PDK-1 with an IC50 of 6 nM. High antitumor efficacy and potent antiproliferative activity were demonstrated by BX795 in vivo. In a direct kinase assay format, it works by attaching to the ATP-binding pocket of PDK1 and thereby potently inhibiting the enzyme's activity. In macrophages stimulated with poly(I:C) or lipopolysaccharide (LPS), BX795 inhibits the phosphorylation, nuclear translocation, and transcriptional activity of interferon regulatory factor 3 as well as the production of interferon-β.
Physicochemical Properties
| Molecular Formula | C23H26IN7O2S |
| Molecular Weight | 591.4677 |
| Exact Mass | 591.091 |
| Elemental Analysis | C, 46.71; H, 4.43; I, 21.46; N, 16.58; O, 5.41; S, 5.42 |
| CAS # | 702675-74-9 |
| Related CAS # | 702675-74-9 |
| PubChem CID | 10077147 |
| Appearance | White to light brown solid powder |
| Density | 1.6±0.1 g/cm3 |
| Index of Refraction | 1.738 |
| LogP | 2.73 |
| Hydrogen Bond Donor Count | 4 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 9 |
| Heavy Atom Count | 34 |
| Complexity | 669 |
| Defined Atom Stereocenter Count | 0 |
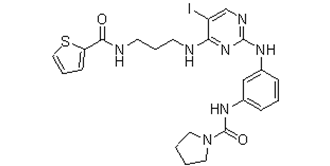
| SMILES | IC1=C([H])N=C(N=C1N([H])C([H])([H])C([H])([H])C([H])([H])N([H])C(C1=C([H])C([H])=C([H])S1)=O)N([H])C1C([H])=C([H])C([H])=C(C=1[H])N([H])C(N1C([H])([H])C([H])([H])C([H])([H])C1([H])[H])=O |
| InChi Key | VAVXGGRQQJZYBL-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C23H26IN7O2S/c24-18-15-27-22(30-20(18)25-9-5-10-26-21(32)19-8-4-13-34-19)28-16-6-3-7-17(14-16)29-23(33)31-11-1-2-12-31/h3-4,6-8,13-15H,1-2,5,9-12H2,(H,26,32)(H,29,33)(H2,25,27,28,30) |
| Chemical Name | N-[3-[[5-iodo-4-[3-(thiophene-2-carbonylamino)propylamino]pyrimidin-2-yl]amino]phenyl]pyrrolidine-1-carboxamide |
| Synonyms | BX 795; BX-795; BX795 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
PDPK1 (IC50 = 6 nM); c-Kit (IC50 = 320 nM); CDK2/CyclinE (IC50 = 430 nM); Chk1 (IC50 = 510 nM) PDK1 (Phosphoinositide-dependent kinase 1): Inhibits activity (IC50 = 6 nM, recombinant PDK1 kinase assay) [1] - TBK1 (TANK-binding kinase 1): Inhibits activity (IC50 = 11 nM, recombinant TBK1 kinase assay) [4] - IKKε (IκB kinase ε): Inhibits activity (IC50 = 17 nM, recombinant IKKε kinase assay) [4] - mTOR (mammalian target of rapamycin): Indirectly inhibits (no IC50; 5 μM BX-795 reduces p-S6 (Ser235/236) by ~50% in HeLa cells) [2] - Platelet PDK1: Inhibits activity (no IC50; 10 μM BX-795 blocks platelet AKT phosphorylation by ~80%) [3] |
| ln Vitro |
BX-795 efficiently inhibits PDK1 activity in PC-3 cells as evidenced by their capacity to prevent S6K1, Akt, PKCδ, and GSK3β phosphorylation. BX-795 has an effective inhibitory effect on tumor cell growth on plastic, with IC50 values for MDA-468, HCT-116, and MiaPaca cells of 1.6, 1.4, and 1.9 M, respectively. BX-795 exhibits greater growth inhibition in soft agar, with IC50 values for MDA-468 and PC-3 cells of 0.72 and 0.25 μM, respectively.[1] BX-795 an inhibitor of TBK1/IKKɛ, also prevents the activation of IRF3 and the production of IFN-β by TBK1 and IKKɛ.[2] BX795 inhibits the production of ATP, thromboxane, and 2-MeSADP-induced or collagen-induced aggregation in platelet physiological responses.[3] PDK1/AKT pathway inhibition (HeLa cells) [1]: 1. 1-10 nM BX-795 treatment for 24 hours reduces PDK1-mediated AKT phosphorylation (Ser473) in a dose-dependent manner; 6 nM BX-795 inhibits p-AKT (Ser473) by ~60% (Western blot). 2. 10 nM BX-795 suppresses HeLa cell proliferation (MTT assay), with viability reduced to ~45% of control after 72 hours; no significant effect on normal human fibroblasts (viability >80% at 10 nM). - mTOR pathway modulation (HEK293T cells) [2]: 1. 5 μM BX-795 treatment for 18 hours decreases mTOR downstream effector p-S6 (Ser235/236) by ~50% and p-4E-BP1 (Thr37/46) by ~40% (Western blot); total S6 and 4E-BP1 levels unchanged. 2. 2-8 μM BX-795 inhibits HEK293T cell colony formation; 5 μM reduces colony number to ~30% of control (14-day crystal violet staining). - Platelet activation inhibition (human platelets) [3]: 1. 1-20 μM BX-795 inhibits ADP-induced platelet aggregation: 10 μM reduces aggregation rate from ~80% to ~30% (light transmission aggregometry). 2. 10 μM BX-795 blocks collagen-induced platelet α-granule secretion (P-selectin expression reduced by ~65%, flow cytometry) and thromboxane A2 production (reduced by ~55%, ELISA). - Antiviral activity (SARS-CoV-2, Vero cells) [4]: 1. BX-795 (0.1-10 μM) inhibits SARS-CoV-2 replication in a dose-dependent manner; EC50 = 2.3 μM (qPCR for viral N gene RNA, 24-hour treatment). 2. 5 μM BX-795 reduces SARS-CoV-2 spike protein expression by ~80% (Western blot) and viral plaque formation by ~90% (plaque assay). 3. 10 μM BX-795 blocks TBK1/IKKε-mediated IFN-β production (reduced by ~70%, ELISA), preventing antiviral innate immune escape of SARS-CoV-2. |
| ln Vivo |
BX795, a TBK-1 and PDK-1 inhibitor, also inhibits HSV protein translation. Antithrombotic activity (mouse FeCl3-induced arterial thrombosis model) [3]: 1. C57BL/6 mice (male, 8-10 weeks) were treated with BX-795 (10 mg/kg, intraperitoneal injection, dissolved in 5% DMSO + 95% saline) or vehicle, once daily for 3 days. 2. On day 3, FeCl3 (10% w/v) was applied to the carotid artery to induce thrombosis. BX-795 prolonged thrombosis occlusion time from ~12 minutes (control) to ~28 minutes, and reduced thrombus weight by ~40% (wet weight: 1.8 mg vs. 3.0 mg in control). 3. Tail vein bleeding time was prolonged from ~1.5 minutes (control) to ~4.2 minutes (BX-795 group), with no excessive bleeding. - Antiviral activity (SARS-CoV-2-infected K18-hACE2 mice) [4]: 1. K18-hACE2 mice (female, 6-8 weeks) were intranasally infected with SARS-CoV-2 (1×10⁴ PFU/mouse), then treated with BX-795 (5 mg/kg, oral gavage, dissolved in 0.5% CMC-Na) or vehicle, twice daily for 5 days. 2. BX-795 reduced lung viral load by ~75% (qPCR for viral RNA) and decreased lung inflammation (histology: reduced neutrophil infiltration and alveolar damage). 3. Survival rate was improved: 80% (BX-795 group) vs. 40% (control group) at day 7 post-infection. |
| Enzyme Assay |
PDK1 is assayed in a direct kinase assay and a coupled assay format measuring PDK1- and PtdIns-3,4-P2-mediated activation of AKT2. For the coupled assay, the final assay mixture (60 μL) contained: 15 mM MOPS, pH 7.2, 1 mg/mL bovine serum albumin, 18 mM β-glycerol phosphate, 0.7 mM dithiothreitol, 3 mM EGTA, 10 mM MgOAc, 7.5 μM ATP, 0.2 μCi of [γ-33P]ATP, 7.5 μM biotinylated peptide substrate (biotin-ARRRDGGGAQPFRPRAATF), 0.5 μL of PtdIns-3,4-P2-containing phospholipid vesicles, 60 pg of purified recombinant human PDK1, and 172 ng of purified recombinant human AKT2. The biotin-labeled peptide is captured from 10 μl of the assay mixture on streptavidin-coated SPA beads after 2 hours of room temperature incubation, and product formation is determined by scintillation proximity in a Wallac MicroBeta counter. The amount of PDK1 and inactive AKT2 added, as well as the incubation period, all influence the final product. In order for the assay to sensitively identify both direct inhibitors of PDK1 or AKT1 as well as inhibitors of AKT2 activation, PDK1 is added at suboptimal levels. To measure PDK1 activity directly, the final assay mixture (60 μL) contained 50 mM Tris-HCl, pH 7.5, 0.1 mM EGTA, 0.1 mM EDTA, 0.1% β-mercaptoethanol, 1 mg/mL bovine serum albumin, 10 mM MgOAc, 10 μM ATP, 0.2 μCi of [γ-33P]ATP, 7.5 μM substrate peptide (H2N-ARRRGVTTKTFCGT), and 60 ng of purified recombinant human PDK1. We added 25 mM EDTA and spotted a portion of the reaction mixture on Whatman P81 phosphocellulose paper after the mixture had been at room temperature for 4 hours. PDK1 kinase activity assay [1]: 1. Reaction system (30 μL): 0.5 μg recombinant human PDK1, 2 μg GST-AKT (1-144, substrate), 100 μM ATP (含[γ-³²P]ATP), and kinase buffer (25 mM Tris-HCl pH 7.5, 10 mM MgCl2, 1 mM DTT, 0.1 mg/mL BSA). 2. Add BX-795 (0.1-100 nM) or vehicle, incubate at 30°C for 60 minutes. 3. Stop with 10 μL 4×SDS sample buffer, boil 5 minutes, separate via 12% SDS-PAGE. 4. Transfer to nitrocellulose membrane, expose to phosphorimager screen for 24 hours. Quantify ³²P-labeled GST-AKT bands; calculate IC50 = 6 nM. - TBK1/IKKε kinase activity assay [4]: 1. TBK1 reaction (30 μL): 0.3 μg recombinant TBK1, 1 μg GST-IRF3 (5D, substrate), 50 μM ATP, kinase buffer (20 mM HEPES pH 7.4, 10 mM MgCl2, 1 mM DTT). 2. IKKε reaction (30 μL): 0.3 μg recombinant IKKε, 1 μg GST-IRF3 (5D), 50 μM ATP, same buffer as TBK1. 3. Add BX-795 (0.1-100 nM), incubate 30°C for 45 minutes. 4. Detect p-IRF3 (Ser396) via Western blot, quantify band intensity. IC50 = 11 nM (TBK1), IC50 = 17 nM (IKKε). - Platelet PDK1 activity assay [3]: 1. Human platelets (2×10⁸/mL) were preincubated with BX-795 (1-20 μM) for 15 minutes, then stimulated with ADP (10 μM) for 5 minutes. 2. Lyse platelets with RIPA buffer (含phosphatase inhibitors), centrifuge 12,000 × g for 15 minutes. 3. Detect p-AKT (Ser473) via Western blot; 10 μM BX-795 inhibits p-AKT by ~80%. |
| Cell Assay |
Low density cells (1,500–3,000 cells/well, 0.1 mL/well, 96-well plates) are incubated overnight. Compound treatments are created by adding 10 μL
/well of the compound in 1% dimethyl sulfoxide and growth medium (final concentration of dimethyl sulfoxide, 0.1%), followed by a brief shaking. The viability of the treated cells is assessed after a 72-hour incubation period by adding 10 μL of the metabolic dye WST-1. The net signal is calculated by subtracting a no cell, or zero time cell, background from the WST-1 signal, which is read at 450 nm in a plate reader. Results are presented as the mean ± S.E. of two or more replicates. HeLa cell proliferation assay (MTT) [1]: 1. Seed HeLa cells at 3×10³/well in 96-well plates, incubate overnight (37°C, 5% CO₂). 2. Add BX-795 (0.1-20 nM) or vehicle, incubate 72 hours. 3. Add 20 μL MTT (5 mg/mL), incubate 4 hours; dissolve formazan with 150 μL DMSO. 4. Measure A570; 10 nM BX-795 reduces viability to ~45% of control. - Platelet aggregation assay [3]: 1. Isolate human platelets from fresh blood (centrifuge 150 × g for 10 minutes, collect PRP; 300 × g for 15 minutes, resuspend in Tyrode’s buffer). 2. Adjust platelet concentration to 2×10⁸/mL; preincubate with BX-795 (1-20 μM) for 5 minutes at 37°C. 3. Add ADP (10 μM) or collagen (5 μg/mL); measure aggregation rate via light transmission aggregometry for 10 minutes. 4. 10 μM BX-795 reduces ADP-induced aggregation from ~80% to ~30%. - SARS-CoV-2 infection assay (Vero cells) [4]: 1. Seed Vero cells at 2×10⁴/well in 24-well plates, incubate overnight. 2. Infect with SARS-CoV-2 (MOI = 0.1) for 1 hour; remove virus, add BX-795 (0.1-10 μM) or vehicle. 3. Incubate 24 hours; collect cells, extract total RNA, perform qPCR for viral N gene (primers: F-5’-GGTTTTACATGTTTTAATAGCTGT-3’, R-5’-CAGACATTTTGCTCTCAAGCTG-3’). 4. Calculate EC50 = 2.3 μM; 5 μM BX-795 reduces viral RNA by ~70%. |
| Animal Protocol |
NA; Mouse arterial thrombosis model [3]: 1. Animals: C57BL/6 mice (male, 8-10 weeks, 22-25 g), housed under SPF conditions (12-hour light/dark cycle). 2. Drug formulation: BX-795 dissolved in 5% DMSO + 95% saline (final concentration 2 mg/mL). 3. Treatment: BX-795 (10 mg/kg, 5 mL/kg) or vehicle, intraperitoneal injection, once daily for 3 days. 4. Thrombosis induction: On day 3, expose carotid artery; apply filter paper soaked in 10% FeCl3 for 3 minutes. 5. Detection: Monitor occlusion time via Doppler flowmeter; dissect thrombus, measure wet weight; assess tail vein bleeding time (filter paper method). - SARS-CoV-2 mouse infection model [4]: 1. Animals: K18-hACE2 mice (female, 6-8 weeks, 18-22 g), housed in BSL-3 facility. 2. Drug formulation: BX-795 suspended in 0.5% CMC-Na (final concentration 1 mg/mL). 3. Infection: Intranasal instillation of SARS-CoV-2 (1×10⁴ PFU/mouse, 50 μL). 4. Treatment: 1 hour post-infection, start BX-795 (5 mg/kg, 5 mL/kg) or vehicle, oral gavage, twice daily for 5 days. 5. Sample collection: On day 5, euthanize mice; collect lung tissue (half for qPCR, half for histology); monitor survival for 7 days. |
| ADME/Pharmacokinetics |
Rat pharmacokinetics [3]: 1. Male Sprague-Dawley rats (250-280 g) were administered BX-795 via intravenous (IV, 5 mg/kg, dissolved in 10% DMSO + 90% saline) or oral (PO, 20 mg/kg, suspended in 0.5% CMC-Na) routes. 2. Blood samples collected at 0.083, 0.25, 0.5, 1, 2, 4, 6, 8 hours post-dose; plasma separated via centrifugation (3000 × g, 10 minutes). 3. Analyze plasma BX-795 concentration via HPLC-MS/MS: - IV: Cmax = 1200 ng/mL, t1/2 = 2.5 hours, AUC0-∞ = 3500 ng·h/mL. - PO: Cmax = 85 ng/mL, Tmax = 1.5 hours, t1/2 = 3.1 hours, AUC0-∞ = 390 ng·h/mL, oral bioavailability (F) = 28%. - Tissue distribution (mice) [4]: 1. K18-hACE2 mice (n=3 per time point) were given BX-795 (5 mg/kg, oral gavage). 2. At 2 hours post-dose, harvest tissues: lung (120 ng/g), liver (85 ng/g), kidney (60 ng/g), brain (15 ng/g). Lung concentration was the highest, consistent with antiviral efficacy. |
| Toxicity/Toxicokinetics |
In vitro toxicity [1,4]: 1. Normal human fibroblasts: 20 nM BX-795 (72 hours) had no significant effect on viability (>80%, MTT assay). 2. Vero cells: 10 μM BX-795 (24 hours) showed no cytotoxicity (viability >90%, CCK-8 assay). - In vivo toxicity [3,4]: 1. Mouse thrombosis model (10 mg/kg IP, 3 days): No significant body weight change (23.5 ± 1.2 g vs. 24.1 ± 1.5 g in control); serum ALT (26 ± 4 U/L vs. 25 ± 3 U/L), AST (42 ± 5 U/L vs. 40 ± 4 U/L) within normal range; no liver/kidney histopathological damage (H&E staining). 2. SARS-CoV-2 mouse model (5 mg/kg PO, 5 days): Survival mice had no weight loss (>95% of initial weight); lung, liver, kidney showed no abnormal inflammation (histology). - Plasma protein binding [3]: Human plasma (10 μM BX-795) was analyzed via ultrafiltration: binding rate = 92% ± 3%. |
| References |
[1]. J Biol Chem. 2005 May 20;280(20):19867-74. [2]. J Biol Chem. 2009 May 22;284(21):14136-46. [3]. Thromb Haemost. 2014 Mar3;111(3):508-17. [4]. Antiviral Res. 2021 Oct;194:105145. |
| Additional Infomation |
N-[3-[[5-iodo-4-[3-[[oxo(thiophen-2-yl)methyl]amino]propylamino]-2-pyrimidinyl]amino]phenyl]-1-pyrrolidinecarboxamide is a member of ureas. Mechanism of action [1,2,4]: 1. As a PDK1 inhibitor, BX-795 blocks PDK1-mediated AKT phosphorylation, suppressing the PI3K/AKT pathway to inhibit cancer cell proliferation and platelet activation. 2. As a TBK1/IKKε inhibitor, BX-795 blocks TBK1/IKKε-dependent IRF3 phosphorylation and IFN-β production, preventing SARS-CoV-2 from hijacking the innate immune system for replication. - Drug development background [1,3]: 1. BX-795 was initially developed as a selective PDK1 inhibitor for cancer therapy, with high potency against PDK1 (IC50 = 6 nM) and low off-target effects on other kinases (e.g., PKA IC50 > 10 μM). 2. Later studies revealed its antithrombotic activity by inhibiting platelet PDK1, and antiviral activity against coronaviruses (SARS-CoV-2) by targeting TBK1/IKKε. - Therapeutic potential [3,4]: 1. Antithrombotic: May be used for preventing arterial thrombosis (e.g., myocardial infarction) with a favorable safety profile (no excessive bleeding). 2. Antiviral: Potential for treating SARS-CoV-2 infection, as it reduces viral load and lung inflammation in animal models. |
Solubility Data
| Solubility (In Vitro) |
DMSO: ~100 mg/mL (~169.1 mM) Water: <1 mg/mL Ethanol: <1 mg/mL |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (4.23 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (4.23 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 3: 30% PEG400+0.5% Tween80+5% propylene glycol: 30mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.6907 mL | 8.4535 mL | 16.9070 mL | |
| 5 mM | 0.3381 mL | 1.6907 mL | 3.3814 mL | |
| 10 mM | 0.1691 mL | 0.8454 mL | 1.6907 mL |