Physicochemical Properties
| Molecular Formula | C25H24N6O3 |
| Molecular Weight | 456.50 |
| Exact Mass | 456.19 |
| Elemental Analysis | C, 65.78; H, 5.30; N, 18.41; O, 10.51 |
| CAS # | 1858206-76-4 |
| Related CAS # | 1858206-76-4 |
| PubChem CID | 118649285 |
| Appearance | White to off-white solid powder |
| LogP | 2.5 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 5 |
| Rotatable Bond Count | 5 |
| Heavy Atom Count | 34 |
| Complexity | 805 |
| Defined Atom Stereocenter Count | 1 |
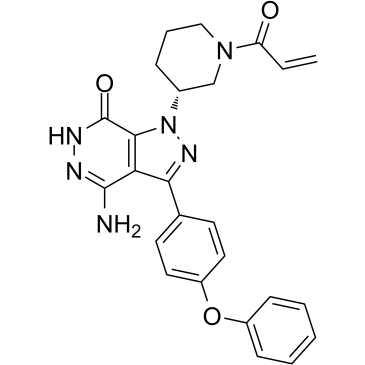
| SMILES | C=CC(=O)N1CCC[C@H](C1)N2C3=C(C(=N2)C4=CC=C(C=C4)OC5=CC=CC=C5)C(=NNC3=O)N |
| InChi Key | QUYXPVXTKPTPCA-QGZVFWFLSA-N |
| InChi Code | InChI=1S/C25H24N6O3/c1-2-20(32)30-14-6-7-17(15-30)31-23-21(24(26)27-28-25(23)33)22(29-31)16-10-12-19(13-11-16)34-18-8-4-3-5-9-18/h2-5,8-13,17H,1,6-7,14-15H2,(H2,26,27)(H,28,33)/t17-/m1/s1 |
| Chemical Name | 4-amino-3-(4-phenoxyphenyl)-1-[(3R)-1-prop-2-enoylpiperidin-3-yl]-6H-pyrazolo[3,4-d]pyridazin-7-one |
| Synonyms | QQN06764; QQN-06764; QQN 06764; BTK-IN-17; BTK-IN-8 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
BTK (IC50 = 2.1 nM) Bruton's Tyrosine Kinase (BTK) with an IC50 value of 1.8 nM for recombinant human BTK enzyme activity; no significant inhibition of other kinases (EGFR, ITK, JAK3) with IC50 > 1000 nM [1] |
| ln Vitro |
BTK inhibitor 17 (compound 8) could form an HB network with hinge key residues Met477, Glu475, and gatekeeper Thr474 by covalently binding to Cys481[1]. BTK inhibitor 17 irreversibly inhibited recombinant human BTK enzyme activity in a dose-dependent manner, with an IC50 of 1.8 nM. Mass spectrometry confirmed covalent binding to the Cys481 residue in the BTK active site [1] - In BTK-dependent B-cell lines (Ramos, Raji), BTK inhibitor 17 suppressed cell proliferation with IC50 values of 23 nM and 31 nM, respectively. Flow cytometry analysis showed induction of early apoptosis (Annexin V-positive/PI-negative) in 35% of Ramos cells after 48-hour treatment with 100 nM [1] - Western blot assays revealed that BTK inhibitor 17 (10–100 nM) dose-dependently reduced BTK phosphorylation (p-BTK Y223) and downstream signaling molecules (p-ERK1/2, p-PLCγ2) in Ramos cells, without affecting total BTK protein levels [1] - Kinase selectivity panel screening showed BTK inhibitor 17 exhibited >500-fold selectivity for BTK over 45 other kinases (e.g., EGFR, ITK, JAK3, SRC), with IC50 values >1000 nM for off-target kinases [1] |
| ln Vivo |
BTK inhibitor 17 (compound 8; 3-10 mg/kg; oral gavage; daily; for 28 days) treatment prevents the disease from progressing significantly, shows a dose-dependent reduction in paw clinical scores, and does not cause a significant reduction in body weight at any dosage[1]. BTK inhibitor 17 (compound 8) exhibits >95% binding to plasma proteins in three different species: mice, rats, and humans. The following parameters are measured in two species following an intravenous injection: half-life (0.32-hours in rats; 0.42-hours in mice); clearance (54.6-21.3 mL/min/kg in rats; 0.82 mL/kg in mice); volume of distribution (1.55-255 l/kg in rats; 0.82 l/kg in mice); and AUC exposure (604 ng.h/mL in rats; 576 ng.h/mL in mice). BTK inhibitor 17 has a better oral bioavailability (rat, 23.7%; mice, 11.2%) and higher Cmax (rat, 466 ng/mL; mice, 252 ng/mL) and plasma exposure (rat, 642 ng.h/mL; mice, 128 ng.h/mL) after oral administration[1]. In a Ramos cell xenograft mouse model (BALB/c nude mice), oral administration of BTK inhibitor 17 (25 mg/kg/day and 50 mg/kg/day for 21 days) resulted in tumor growth inhibition (TGI) rates of 62% and 85%, respectively, compared to vehicle controls [1] - BTK inhibitor 17 treatment (50 mg/kg, oral) significantly reduced p-BTK Y223 levels in tumor tissues by 78% (western blot) and decreased Ki-67-positive proliferating cells by 60% (immunohistochemistry) [1] - No significant body weight loss or obvious toxicity signs were observed in treated mice during the 21-day study period [1] |
| Enzyme Assay |
BTK enzyme activity assay: Recombinant human BTK protein was incubated with serial dilutions of BTK inhibitor 17 (0.01–100 nM) at 37°C for 1 hour. ADP-Glo kinase assay system was used to measure ATP consumption, with a peptide substrate specific for BTK. Luminescence intensity was detected to quantify enzyme activity, and IC50 values were calculated from dose-response curves [1] - Covalent binding validation assay: BTK inhibitor 17 was incubated with recombinant BTK at 37°C for 2 hours. The mixture was digested with trypsin, and the resulting peptides were analyzed by LC-MS/MS. Mass shifts of the peptide containing Cys481 were used to confirm covalent modification [1] - Kinase selectivity assay: BTK inhibitor 17 (1 μM) was screened against a panel of 46 kinases. Enzyme activity was measured using kinase-specific substrates and detection systems. Inhibition rates <20% were considered non-significant, and IC50 values were determined for kinases with inhibition rates >50% [1] |
| Cell Assay |
Animal Model: Male Balb/C mice injected with collagen Dosage: 3 mg/kg or 10 mg/kg Administration: Oral gavage; daily; for 28 days Result: Inhibited the significant progression of the disease and exhibited a clear dose-dependent reduction per paw clinical scores. Cell proliferation assay: Ramos and Raji cells were seeded in 96-well plates (3×10³ cells/well) and treated with BTK inhibitor 17 (0.1–1000 nM) for 72 hours. Cell viability was measured by CCK-8 assay, and IC50 values were calculated using GraphPad Prism software [1] - Apoptosis assay: Ramos cells (1×10⁶ cells/mL) were treated with BTK inhibitor 17 (10–1000 nM) for 48 hours. Cells were stained with Annexin V-FITC and PI, then analyzed by flow cytometry to quantify early and late apoptotic populations [1] - Western blot assay: Ramos cells were treated with BTK inhibitor 17 for 24 hours, then lysed in RIPA buffer. Protein extracts were separated by SDS-PAGE, transferred to membranes, and probed with antibodies against p-BTK Y223, total BTK, p-ERK1/2, p-PLCγ2, and β-actin. Immunoreactive bands were quantified by densitometry [1] |
| Animal Protocol |
Ramos xenograft model: BALB/c nude mice (6-week-old) were subcutaneously inoculated with 5×10⁶ Ramos cells. When tumors reached 100–150 mm³, mice were randomly divided into three groups (n=6): vehicle, 25 mg/kg BTK inhibitor 17, and 50 mg/kg BTK inhibitor 17. The compound was suspended in 0.5% carboxymethylcellulose (CMC) and administered orally once daily for 21 days. Tumor volume and body weight were measured every 3 days [1] - Tumor tissue analysis: At the end of the study, mice were euthanized, and tumor tissues were collected. Half of each tumor was fixed in formalin for immunohistochemistry (Ki-67 staining), and the other half was frozen for western blot analysis of p-BTK and downstream signaling molecules [1] |
| ADME/Pharmacokinetics |
Oral bioavailability: BTK inhibitor 17 showed 42% oral bioavailability in SD rats after a 10 mg/kg oral dose. Intravenous administration (5 mg/kg) resulted in a plasma Cmax of 892 ng/mL, while oral administration (10 mg/kg) yielded a Cmax of 321 ng/mL and Tmax of 1.5 hours [1] - Elimination and distribution: The plasma elimination half-life (t1/2) was 6.8 hours in rats. The compound showed good tissue penetration, with tumor-to-plasma concentration ratio of 2.3 in xenograft mice. Approximately 55% of the drug was excreted via feces and 30% via urine within 48 hours [1] - Metabolic stability: BTK inhibitor 17 exhibited high stability in human liver microsomes (t1/2 > 2 hours) and was minimally metabolized by CYP450 isoforms (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4) [1] |
| Toxicity/Toxicokinetics |
Acute toxicity: No mortality or severe toxicity was observed in ICR mice treated with single oral doses of BTK inhibitor 17 up to 200 mg/kg. Mild diarrhea was observed at 100 mg/kg but resolved within 48 hours [1] - Subchronic toxicity: SD rats administered BTK inhibitor 17 (50 mg/kg/day, oral) for 28 days showed no significant changes in hematology, serum biochemistry (ALT, AST, BUN, creatinine), or organ weights. Plasma protein binding rate was 88% [1] |
| References |
[1]. Discovery and Evaluation of Pyrazolo[3,4- d]pyridazinone as a Potent and Orally Active Irreversible BTK Inhibitor. ACS Med Chem Lett. 2019 Dec 11;11(10):1863-1868. |
| Additional Infomation |
BTK inhibitor 17 is an irreversible BTK inhibitor with a pyrazolo[3,4-d]pyridazinone scaffold, designed to covalently bind to the Cys481 residue of BTK [1] - The compound exerts its antitumor effect by inhibiting BTK-mediated B-cell receptor (BCR) signaling, leading to suppressed proliferation and induced apoptosis of B-cell malignant cells [1] - BTK inhibitor 17 exhibits favorable oral bioavailability, metabolic stability, and kinase selectivity, making it a potential candidate for the treatment of B-cell malignancies (e.g., non-Hodgkin lymphoma) [1] |
Solubility Data
| Solubility (In Vitro) | DMSO: ~100 mg/mL (~219.1 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.48 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.48 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (5.48 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1906 mL | 10.9529 mL | 21.9058 mL | |
| 5 mM | 0.4381 mL | 2.1906 mL | 4.3812 mL | |
| 10 mM | 0.2191 mL | 1.0953 mL | 2.1906 mL |