Branebrutinib (formerly BMS-986195) is a highly selective and rapidly acting covalent/irreversible inhibitor of Bruton’s Tyrosine Kinase (BTK) with IC50 of<1 nM and robust efficacy at low doses in preclinical models of RA (Rheumatoid Arthritis) and Lupus Nephritishighly. BMS-986195 functions by covalently altering a cysteine residue located in the active site of BTK. Its selectivity varies from 9 to 1010 fold within the Tec family, and it is more than 5000 times selective for BTK over all other kinases. BMS-986195 rapidly inactivated BTK in human whole blood (3.5×10-4 nM-1·min-1) and potently inhibited B cell proliferation, CD86 expression, and antigen-dependent interleukin-6 production (IC50 <1 nM) without affecting antigen-independent measures in the same cells. Comparable efficacy was assessed in relation to FcγR-dependent TNF-α synthesis in human cells. After just the second dose, mice receiving a dose as low as 0.5 mg/kg orally (PO) daily (QD) had a 98% peak BTK inactivation. BTK was dose-dependently inactivated to comparable levels in the spleen, lymph nodes, and whole blood. In murine models of RA, such as CIA and CAIA, BMS-986195 showed strong efficacy in preventing clinically apparent disease, histologic joint damage, and loss of bone mineral density. The highest level of effectiveness was noted in both models at doses ≤0.5 mg/kg PO QD, which resulted in ≥95% inactivation of BTK in vivo. In the NZB/W mouse model of lupus, the compound was also highly protective against nephritis at similar doses. A single or multiple doses of BMS-986195 were administered to cynomolgus monkeys in order to study the dynamics of BTK inactivation and resynthesis. With a single dosage of BMS-986195 at 0.5 mg/kg PO, 100% peak inactivation of BTK was attained.
Physicochemical Properties
| Molecular Formula | C20H23FN4O2 | |
| Molecular Weight | 370.43 | |
| Exact Mass | 370.18 | |
| Elemental Analysis | C, 64.85; H, 6.26; F, 5.13; N, 15.13; O, 8.64 | |
| CAS # | 1912445-55-6 | |
| Related CAS # |
|
|
| PubChem CID | 121293929 | |
| Appearance | White to off-white solid powder | |
| LogP | 2.8 | |
| Hydrogen Bond Donor Count | 3 | |
| Hydrogen Bond Acceptor Count | 4 | |
| Rotatable Bond Count | 3 | |
| Heavy Atom Count | 27 | |
| Complexity | 657 | |
| Defined Atom Stereocenter Count | 1 | |
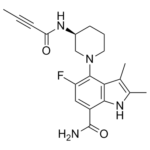
| SMILES | FC1C=C(C(N)=O)C2=C(C(C)=C(C)N2)C=1N1CCC[C@@H](C1)NC(C#CC)=O |
|
| InChi Key | VJPPLCNBDLZIFG-ZDUSSCGKSA-N | |
| InChi Code | InChI=1S/C20H23FN4O2/c1-4-6-16(26)24-13-7-5-8-25(10-13)19-15(21)9-14(20(22)27)18-17(19)11(2)12(3)23-18/h9,13,23H,5,7-8,10H2,1-3H3,(H2,22,27)(H,24,26)/t13-/m0/s1 | |
| Chemical Name | 4-[(3S)-3-(but-2-ynoylamino)piperidin-1-yl]-5-fluoro-2,3-dimethyl-1H-indole-7-carboxamide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
BTK (IC50 = 0.1 nM); TEC (IC50 = 0.9 nM); BMX (IC50 = 1.5 nM); TXK (IC50 = 5 nM)
Bruton’s Tyrosine Kinase (BTK) (Ki = 0.3 nM for human BTK; IC₅₀ = 0.5 nM for human BTK kinase activity; IC₅₀ = 2.1 nM for BTK autophosphorylation in Ramos cells); >1000-fold selectivity over ITK (IC₅₀ = 650 nM), EGFR (IC₅₀ > 1000 nM), ERBB2 (IC₅₀ > 1000 nM), JAK2 (IC₅₀ > 1000 nM), and >500-fold selectivity over BMX (IC₅₀ = 280 nM), TEC (IC₅₀ = 320 nM) [2] |
|
| ln Vitro |
|
|
| ln Vivo |
|
|
| Enzyme Assay |
BMS-986195 has proven to be highly effective in mitigating clinically apparent disease, histologic joint damage, and loss of bone mineral density in mice in murine models of RA, such as CIA and CAIA. The highest level of effectiveness is seen in both mice and monkeys at doses ≤0.5 mg/kg PO QD, which results in ≥95% inactivation of BTK in vivo. In the NZB/W mouse model of lupus, BMS-986195 is also highly protective against nephritis at similar doses. Single or multiple doses of BMS-986195 are administered to cynomolgus monkeys in order to study the dynamics of BTK inactivation and resynthesis. With a single dose of BMS-986195 at 0.5 mg/kg PO, 100% peak inactivation of BTK is achieved[1]. BTK kinase activity assay (HTRF): Recombinant human BTK kinase domain is mixed with ATP (Km concentration), biotinylated peptide substrate, and serially diluted BMS-986195 (0.001–1000 nM) in reaction buffer. The mixture is incubated at 30°C for 60 minutes, then stopped by adding streptavidin-europium cryptate and anti-phosphotyrosine antibody-XL665. Fluorescence resonance energy transfer (FRET) signal is measured at 665 nm/620 nm, and IC₅₀ values are calculated via nonlinear regression. Time-dependent inhibition is assessed by varying incubation times (15–120 minutes) [2] - BTK covalent binding assay (LC-MS): Recombinant human BTK is incubated with BMS-986195 (10 nM) for 0–120 minutes, then digested with trypsin. The digest is analyzed by liquid chromatography-mass spectrometry (LC-MS) to detect the covalent adduct between BMS-986195 and the Cys481-containing peptide, confirming irreversible binding [2] - Kinase selectivity assay: A panel of 468 human kinases is screened with BMS-986195 at 100 nM. Kinase activity is measured using radiometric or fluorescent assays, and selectivity scores (S₁₀, S₃₀) are calculated to assess off-target kinase inhibition [2] |
|
| Cell Assay |
BMS-986195 effectively deactivates BTK in human whole blood at a speed of 3.5×10-4nM-1•min-1. It also significantly suppresses antigen-dependent interleukin-6 production, CD86 expression, and B cell proliferation (IC50<1 nM) in B cells, while having no effect on antigen-independent measures in them. Comparable efficacy is assessed in relation to FcγR-dependent TNF-α synthesis in human cells. BTK/PLCγ2 phosphorylation western blot assay: Ramos or Raji cells are seeded in 6-well plates (2×10⁶ cells/well) and incubated overnight. Cells are pretreated with BMS-986195 (0.01–100 nM) for 1 hour, then stimulated with anti-IgM (10 μg/mL) for 5 minutes. Cells are lysed in RIPA buffer with protease/phosphatase inhibitors, and proteins are analyzed by western blot using antibodies against phospho-BTK (Y223), total BTK, phospho-PLCγ2 (Y759), total PLCγ2, and GAPDH (loading control) [2] - B cell proliferation assay (CFSE): Primary human B cells or Ramos cells are labeled with carboxyfluorescein succinimidyl ester (CFSE), seeded in 96-well plates (1×10⁵ cells/well), and pretreated with BMS-986195 (0.01–100 nM) for 1 hour. Cells are stimulated with anti-IgM (10 μg/mL) + IL-4 (20 ng/mL) for 72 hours, then analyzed by flow cytometry to measure CFSE dilution (proliferation index) [2] - Cytokine secretion assay (ELISA): Primary human B cells are seeded in 24-well plates (2×10⁶ cells/well), pretreated with BMS-986195 (0.1–100 nM) for 1 hour, then stimulated with LPS (1 μg/mL) for 24 hours. Culture supernatants are collected, and IL-6, TNF-α, and CXCL10 levels are measured by ELISA. Inhibition rates are calculated relative to vehicle controls [1][2] |
|
| Animal Protocol |
NZB/W lupus-prone mouse model 0.2, 0.5, and 1.5 mg/kg by oral gavage Mouse CIA model study: DBA/1J mice (6–8 weeks old, n=8 per group) are immunized subcutaneously with bovine type II collagen emulsified in complete Freund's adjuvant on day 0 and day 21. BMS-986195 is dissolved in 0.5% methylcellulose and administered orally at doses of 0.3, 1, 3 mg/kg once daily from day 14 to day 28. Vehicle group receives 0.5% methylcellulose. Clinical scores (swelling, redness, joint function) are assessed daily. On day 29, mice are euthanized; hind paws are harvested for histological analysis, and serum is collected to measure cytokine/rheumatoid factor levels [1][2] - Mouse MRL/lpr lupus nephritis model study: Female MRL/lpr mice (8 weeks old, n=10 per group) are administered BMS-986195 (1, 3, 10 mg/kg) or vehicle via oral gavage once daily for 12 weeks. Proteinuria is measured weekly using a urine protein assay kit. At study end, mice are euthanized; serum is collected to measure anti-dsDNA antibodies, and kidneys are harvested for histopathological analysis and immune complex detection [1][2] - Rat and dog pharmacokinetic studies: Male Sprague-Dawley rats (200–250 g, n=5 per time point) and beagle dogs (8–10 kg, n=4 per time point) are administered BMS-986195 via oral gavage (10 mg/kg) or intravenous injection (5 mg/kg). Blood samples are collected at 0.25, 0.5, 1, 2, 4, 8, 12, 24 hours post-dosing. Plasma drug concentrations are measured by LC-MS/MS, and pharmacokinetic parameters are calculated using non-compartmental analysis [2] |
|
| ADME/Pharmacokinetics |
In rats: Oral administration (10 mg/kg) results in peak plasma concentration (Cₘₐₓ) = 2.8 μg/mL, time to Cₘₐₓ (Tₘₐₓ) = 1.0 hour, terminal half-life (t₁/₂) = 7.2 hours, volume of distribution (Vd) = 3.5 L/kg, and oral bioavailability = 70%. Intravenous administration (5 mg/kg) shows clearance (CL) = 0.39 L/h/kg [2] - In dogs: Oral administration (10 mg/kg) results in Cₘₐₓ = 3.2 μg/mL, Tₘₐₓ = 1.2 hours, t₁/₂ = 9.5 hours, Vd = 3.2 L/kg, and oral bioavailability = 78%. Intravenous administration (5 mg/kg) shows CL = 0.28 L/h/kg [2] - Tissue distribution (rats, 2 hours post-oral 10 mg/kg): Preferential distribution to spleen (tissue-to-plasma ratio = 3.8), lymph nodes (3.5), liver (2.9), lung (2.6), kidney (2.3), and joint synovium (2.1); low brain penetration (tissue-to-plasma ratio = 0.4) [2] - Excretion (rats): 72 hours after intravenous administration (5 mg/kg), 65% of the dose is excreted in feces (32% as parent drug, 33% as metabolites) and 25% in urine (10% as parent drug, 15% as metabolites) [2] - Metabolism: Major metabolic pathways in humans include oxidation (CYP3A4-mediated) and glucuronidation; no toxic metabolites are detected in liver microsome studies [2] |
|
| Toxicity/Toxicokinetics |
Plasma protein binding: BMS-986195 has a plasma protein binding rate of 96% in human plasma, 94% in rat plasma, and 95% in dog plasma (measured by ultrafiltration) [2] - Acute toxicity: In rats and dogs, oral LD₅₀ > 300 mg/kg. No overt toxicity (weight loss, convulsions, mortality) is observed at doses up to 150 mg/kg in a 7-day acute study [2] - Subchronic toxicity (28-day repeated oral dosing in rats): Doses of 10, 30, 100 mg/kg/day do not cause significant changes in body weight, food intake, hematological parameters (RBC, WBC, platelets), or liver/kidney function (ALT, AST, creatinine, BUN). No histopathological abnormalities are found in major organs (liver, kidney, heart, lung, spleen, lymph nodes) [2] - Drug-drug interaction: In vitro studies show no inhibition of cytochrome P450 enzymes (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4) at concentrations up to 10 μM; weak induction of CYP3A4 (1.3-fold at 10 μM) is observed [2] |
|
| References |
[1]. BMS-986195 Is a Highly Selective and Rapidly Acting Covalent Inhibitor of Bruton’s Tyrosine Kinase with Robust Efficacy at Low Doses in Preclinical Models of RA and Lupus Nephritis. 2017 ACR/ARHP Annual Meeting, September 18, 2017. [2]. Discovery of Branebrutinib (BMS-986195): A Strategy for Identifying a Highly Potent and Selective Covalent Inhibitor Providing Rapid in Vivo Inactivation of Bruton's Tyrosine Kinase (BTK). J Med Chem. 2019 Apr 11;62(7):3228-3250. |
|
| Additional Infomation |
Branebrutinib is under investigation in clinical trial NCT02705989 (Safety, Tolerability and Relative Bioavailability Study of BMS-986195 in Healthy Subjects). BMS-986195 (Branebrutinib) is a highly potent, selective, orally bioavailable covalent inhibitor of Bruton’s Tyrosine Kinase (BTK), developed for the treatment of autoimmune diseases [2] - Mechanism of action: Irreversibly binds to the cysteine residue (Cys481) in the active site of BTK via a covalent warhead, inhibiting BTK kinase activity and blocking B cell receptor (BCR)-mediated signaling. This suppresses B cell activation, proliferation, and secretion of pro-inflammatory cytokines/autoantibodies, which drive pathogenesis of rheumatoid arthritis (RA) and lupus nephritis [1][2] - Therapeutic potential: Preclinical data support its utility in autoimmune diseases, including RA and lupus nephritis, with robust efficacy at low doses (0.3–10 mg/kg oral) and rapid onset of action (BTK inhibition within 2 hours) [1][2] - Druggability advantages: High oral bioavailability (70–78% in preclinical species), long half-life (7.2–9.5 hours) supporting once-daily dosing, target tissue (spleen, lymph nodes, synovium) distribution, and high BTK selectivity minimizing off-target effects (e.g., ITK inhibition-related T cell dysfunction) [2] - Distinctive features: Covalent binding confers prolonged BTK inhibition (sustained for 24 hours post-dosing) at low doses, reducing dosing frequency and potential toxicity; minimal impact on T cells and myeloid cells preserves normal immune function [1][2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 3.75 mg/mL (10.12 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 37.5 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 3.75 mg/mL (10.12 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 37.5 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6996 mL | 13.4978 mL | 26.9957 mL | |
| 5 mM | 0.5399 mL | 2.6996 mL | 5.3991 mL | |
| 10 mM | 0.2700 mL | 1.3498 mL | 2.6996 mL |