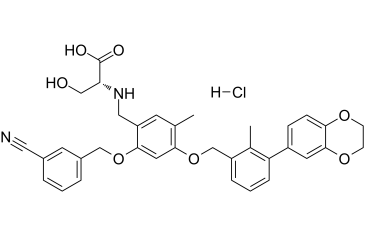
BMS-1001 HCl is a novel and potent small molecule inhibitor of the PD-1/PD-L1 protein protein interaction with IC50 value of 2.25 nM in cell free assays. It was first discovered by Bristol-Myers Squibb. Blockade of the PD-1/PD-L1 immune checkpoint pathway with monoclonal antibodies has provided significant advances in cancer treatment. The antibody-based immunotherapies carry a number of disadvantages such as the high cost of the antibodies, their limited half-life, and immunogenicity. Development of small-molecule PD-1/PD-L1 inhibitors that could overcome these drawbacks is slow because of the incomplete structural information for this pathway. The first chemical PD-1/PD-L1 inhibitors, BMS-1001 and its analogs, have been recently disclosed by Bristol-Myers Squibb.
Physicochemical Properties
| Molecular Formula | C35H35CLN2O7 |
| Molecular Weight | 631.1146 |
| Exact Mass | 630.21327 |
| Elemental Analysis | C, 66.61; H, 5.59; Cl, 5.62; N, 4.44; O, 17.75 |
| CAS # | 2113650-04-5 |
| Related CAS # | 2113650-03-4;2113650-04-5 (HCl); |
| PubChem CID | 145925650 |
| Appearance | White to off-white solid powder |
| Hydrogen Bond Donor Count | 4 |
| Hydrogen Bond Acceptor Count | 9 |
| Rotatable Bond Count | 12 |
| Heavy Atom Count | 45 |
| Complexity | 957 |
| Defined Atom Stereocenter Count | 1 |
| SMILES | Cl[H].O(C([H])([H])C1C([H])=C([H])C([H])=C(C2C([H])=C([H])C3=C(C=2[H])OC([H])([H])C([H])([H])O3)C=1C([H])([H])[H])C1=C([H])C(=C(C([H])=C1C([H])([H])[H])C([H])([H])N([H])[C@@]([H])(C(=O)O[H])C([H])([H])O[H])OC([H])([H])C1C([H])=C([H])C([H])=C(C#N)C=1[H] |
| InChi Key | (2-((3-Cyanobenzyl)oxy)-4-((3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-2-methylbenzyl)oxy)-5-methylbenzyl)-D-serine hydrochloride |
| InChi Code | VKDUZONDVBDCAX-VNUFCWELSA-N SMILES |
| Chemical Name | BMS-1001 HCl BMS 1001 HCl BMS1001 HCl BMS-1001 hydrochloride BMS 1001 hydrochloride BMS1001 hydrochloride |
| Synonyms | BMS-1001 HCl; BMS 1001 HCl; BMS1001 HCl; 2113650-04-5; BMS-1001 hydrochloride; BMS-1001 (hydrochloride); BMS-1001 HCl; (2-((3-Cyanobenzyl)oxy)-4-((3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-2-methylbenzyl)oxy)-5-methylbenzyl)-D-serine hydrochloride; (2R)-2-[[2-[(3-cyanophenyl)methoxy]-4-[[3-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-methylphenyl]methoxy]-5-methylphenyl]methylamino]-3-hydroxypropanoic acid;hydrochloride; (2R)-2-[({2-[(3-cyanophenyl)methoxy]-4-{[3-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-methylphenyl]methoxy}-5-methylphenyl}methyl)amino]-3-hydroxypropanoic acid hydrochloride; N-[[2-[(3-cyanophenyl)methoxy]-4-[[3-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-methylphenyl]methoxy]-5-methylphenyl]methyl]-D-serine, monohydrochloride; BMS-1001 hydrochloride; BMS 1001 hydrochloride; BMS1001 hydrochloride |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment, avoid exposure to moisture. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
PD-1/PD-L1 interaction Human programmed death-ligand 1 (PD-L1) (blocks PD-1/PD-L1 interaction)[2] |
| ln Vitro |
BMS-1001 attaches itself to human PD-L1 and prevents it from interacting with PD-1. BMS-1001 exhibits minimal toxicity towards the cell lines examined and inhibits the communication between soluble PD-L1 and cell surface-expressed PD-1. The inhibitory effect of soluble PD-L1 on T cell receptor-mediated T lymphocyte activation is lessened by BMS-1001 [1]. BMS-1001 binds to human PD-L1 and blocks its interaction with PD-1, as shown by NMR-based antagonist induced dissociation assay (AIDA). It alleviates the inhibitory effect of both soluble and membrane-bound PD-L1 on T-cell receptor (TCR)-mediated activation of Jurkat T cells (PD-1 effector cells). In a PD-1/PD-L1 checkpoint assay using co-culture of PD-1 effector cells and artificial antigen-presenting cells expressing PD-L1, BMS-1001 dose-dependently induced luciferase activity, demonstrating its ability to antagonize the PD-1/PD-L1 checkpoint at the cellular interface, though with lower potency and maximal activation compared to therapeutic antibodies.[2] BMS-1001 dose-dependently abolished the inhibition of T-cell activation by soluble PD-L1 (sPD-L1). At concentrations representing 2- and 5-fold molar excess over sPD-L1, it completely restored cell activation to the level observed with anti-CD3 antibody alone.[2] Flow cytometry analysis showed that pre-incubation of fluorescently labeled sPD-L1 with BMS-1001 significantly decreased its binding to PD-1-expressing Jurkat cells, indicating interference with the PD-1/PD-L1 interaction at the cell surface.[2] BMS-1001 induces dimerization of soluble PD-L1 in solution, as confirmed by NMR titration (linewidth broadening), size exclusion chromatography (shift in retention time), and cross-linking experiments (SDS-PAGE showing dimer band).[2] |
| ln Vivo |
In Vivo Antitumor Activity of NP19 [[an analog of BMS-1001]] in an H22 Hepatoma Mouse Model[3] Encouraged by the excellent in vivo antitumor efficacy of NP19 on the melanoma B16-F10 tumor model, and the fact that PD-1/PD-L1 inhibitors have broad spectrum of antitumor activities, we further evaluated the in vivo antitumor efficacy of compound NP19 using an H22 hepatoma tumor model in BALB/c mice. Each mouse was injected with 0.8 million H22 cells subcutaneously into the right flank. After tumors reached approximately 100 mm3 in volume, mice were randomized and treated by intraperitoneal (i.p.) injection of NP19 or a vehicle solution for 14 days. As shown in Figure 8, NP19 demonstrated significant in vivo antitumor efficacy with a TGI of 76.5% at the dose of 25 mg/kg (Figure 8A, 8B, 8C). In addition, NP19 did not cause an obvious body weight loss (Figure 8D), indicating that the compound was well tolerated. In Vivo Antitumor Activity of NP19 [[an analog of BMS-1001]] in a B16-F10 Mouse Melanoma Model[3] To determine whether the in vitro anti-PD-1/PD-L1 activity of the newly synthesized compounds can be translated into in vivo efficacy, we tested the antitumor activity of compound NP19 on a mice melanoma B16–F10 tumor model. NP19 was chosen for the in vivo efficacy study due to the ease of synthesis and less cytotoxicity (Table 9) when compared to the more potent compound NP2 or equally potent compound NP12. We treated BALB/c mice bearing melanoma tumors with vehicle control and NP19 (25 mg/kg, 50 mg/kg, 100 mg/kg) administered via intragastric gavage once a day for 15 days. As shown in Figure 6, after 15 days of treatment, the growth of melanoma tumors was inhibited dramatically following NP19 treatment. In Vivo Pharmacokinetic Properties of NP19 [[an analog of BMS-1001]][3] As compound NP19 showed high potency in vitro, the pharmacokinetic (PK) profiles were next evaluated in male Sprague–Dawley rats by intravenous and oral administration. The key p.o. and i.v. administration PK parameters are summarized in Table 8. After a single i.v. administration with 1 mg/kg compound NP19, the half time (t1/2), the clearance rate (CL), and the apparent distribution volume (Vss) of NP19 are 1.5 ± 0.5 h, 0.9 ± 0.2 L/h/kg, and 2.1 ± 0.5 L/kg, respectively. When NP19 was administrated by the oral route at 10 mg/kg, the oral absorption (Tmax = 0.6 ± 0.2 h), long half-life (t1/2 = 10.9 ± 7.7 h), and oral bioavailability (F = 5%) were observed. In addition, no apparent adverse effects were observed in rats. NP19 showed a much longer half-life (10.9 h) following oral gavage as compared to the i.v. half-life (1.5 h); this may be due to the high lipophilicity (logP = 7.9) or poor aqueous solubility of NP19. As a result, NP19 exhibited flip-flop pharmacokinetics. Such flip-flop pharmacokinetics can sometimes occur for poorly water-soluble compounds such as Rebamipide, which has a t1/2 (p.o.)/t1/2 (i.v.) ratio of 13.5 due to poor water solubility (7.6 μg/mL). Another example is the lipophilic compound IAT (an antitubulin agent with 19 μg/mL of water solubility) reported by Chien-ming Li et al., which has a t1/2 (p.o.)/t1/2 (i.v.) ratio of ∼5, similar to NP19 [t1/2 (p.o.)/t1/2 (i.v.) = 7.1]. Due to the low oral bioavailability of the compound NP19, we presumed that a high dosage is needed to offer sufficient drug concentration to exhibit antitumor efficacy. Therefore, we further studied the in vivo activity of compound NP19. |
| Enzyme Assay |
In Vitro PD-1/PD-L1 Binding Assay[3] \nThe ability of compounds in inhibition of PD-1/PD-L1 interaction was investigated using a PD-1/PD-L1 homogenous time-resolved fluorescence (HTRF) binding assay. The PD-1/PD-L1 binding assay kits were purchased from Cisbio. The experiments were performed according to the instruction manual which could be downloaded at https://www.cisbio.com/usa/drug-discovery/human-pd1pd-l1-biochemical-interaction-assay.\n \n\n\nBMS disclosed recently the first nonpeptidic small molecule inhibitors against the PD-1/PD-L1 pathway that showed the activity in a homogeneous time-resolved fluorescence (HTRF) binding assay; however no further data supporting their activity were provided. \n\nNMR measurements[2] \nUniform 15N labelling was obtained by expressing proteins in the M9 minimal medium containing 15NH4Cl as the sole nitrogen source. For NMR measurements the buffer was exchanged by gel filtration to PBS pH 7.4 (PD-L1) or 25 mM sodium phosphate containing 100 mM NaCl pH 6.4 (PD-1). 10% (v/v) of D2O was added to the samples to provide the lock signal. All spectra were recorded at 300K using a Bruker Avance III 600 MHz spectrometer. The interaction of the compounds with PD-L1 was evaluated by monitoring the perturbations in chemical shifts of NMR resonances in the 1H-15N 2D HMQC upon titration with the compound. The ability of tested compounds to dissociate PD-L1/PD-1 was evaluated using the Antagonist Induced Dissociation Assay. In brief, 15N-labeled PD-1 (0.2 mM) was slightly overtitrated with the unlabeled PD-L1. The compounds were aliquoted into the resulting mixture. During the experiment the 1H-15N signals were monitored by the HMQC.\n \n\nPD1/PD-L1 checkpoint assay[2] \nThe aAPCs were seeded in white 96-well plates at the density of 10 000 per well in the culture medium 24 h prior to the assay. On the day of the assay, 3.5x serial dilutions of the antibodies were prepared in the RPMI 1640-containing 1% FBS. Serial dilutions of BMS compounds [BMS-1001] were prepared in DMSO and formulated in RPMI 1640-containing 1% FBS. By this, the concentration of DMSO was kept constant in all samples. 95 μl of the medium was removed from the wells and the cells were overlaid with 40 μl of the compound dilutions. 20 000 of ECs were added to each well in 40 μl RPMI 1640 containing 1% FBS. Following 6 h incubation at 37°C, the plates were equilibrated at room temperature for 10 min and 80 μl of the Bio-Glo reagent was added to each well. After incubation for 10 min, luminescence was quantified using FlexStation 3. Half maximal effective concentrations (EC50) and maximal luminescence values (RLUmax) were determined by fitting the Hill equation to the experimental data.\n \nPD-1/sPD-L1 effector assay[2] \nFor the evaluation of the BMS impact on T cell inhibition by soluble PD-L1, the ECs were stimulated with the anti-CD3 antibody in the presence of the recombinant human sPD-L1. For this, the 96-well white flat bottom plates were coated overnight at 4°C with 5 μg/ml of the anti-CD3 antibody or the isotype control solution in PBS. The antibody solution was removed and the plates were washed 3 times with PBS and dried. sPD-L1 (aa 18–134) was diluted in PBS supplemented with the penicillin/streptomycin solution (100 U/ml final concentration each) in the presence of the BMS compounds [BMS-1001] or a corresponding volume of DMSO. Then, 15 μl of the solution was added to each well of the antibody-coated plate. ECs were centrifuged and diluted to 50 000 per ml, and 60 μl of the cell solution was added to each well. The final concentration of sPD-L1 was 10 μg/ml (0.6 μM). The final concentrations of the BMS compounds [BMS-1001] were: 0.12, 0.3, 1.2 and 3 μM, giving the following BMS:sPD-L1 molar ratios: 1:5, 1:2, 2:1 and 5:1. The cells were cultured for 24 h and the luciferase activity assay was performed using the Bio-Glo Luciferase Assay System according to the manufacturer's instructions. The ability of BMS-1001 to disrupt the PD-1/PD-L1 interaction was evaluated using an NMR-based antagonist induced dissociation assay (AIDA). Briefly, ¹⁵N-labeled PD-1 was slightly overtitrated with unlabeled PD-L1 to form a complex. The addition of BMS-1001 to the mixture was monitored by ¹H-¹⁵N HMQC NMR. Dissociation of the complex was visualized by the restoration of sharp ¹H-¹⁵N signals of PD-1, which had been broadened due to complex formation.[2] Direct NMR titration was used to assess the interaction of BMS-1001 with PD-L1. Uniformly ¹⁵N-labeled PD-L1 was titrated with the compound, and changes in chemical shifts and linewidth broadening in the ¹H-¹⁵N HMQC spectra were monitored to confirm binding and indicate compound-induced increase in molecular weight (dimerization).[2] |
| Cell Assay |
Cell lines[2] To verify the potency of BMS compounds [BMS-1001] in the inhibition of the PD-1/PD-L1 interactions, a cell-based model of the PD-1/PD-L1 immune checkpoint blockade was used. In the assay, two model cell lines are utilized: the artificial Antigen-Presenting Cells (PD-L1+ aAPC/CHO-K1 cells, called aAPCs) overexpressing TCR ligand and PD-L1, and T cell surrogate, a modified Jurkat T cell line overexpressing PD-1 and carrying a luciferase reporter under the control of NFAT promoter (PD-1 Effector Cells, called ECs). The cells were obtained from Promega and cultured in the RPMI 1640 medium supplemented with 10% Fetal Bovine Serum, 100 U/ml Penicillin and 100 U/ml Streptomycin. Additionally, the cells were propagated in a constant presence of Hygromycin B (50 μg/ml) and G418 (250 μg/ml) to provide a stable expression of the introduced genetic constructs. The two latter antibiotics were omitted in the experiments. Overexpression of PD-1 on ECs and PD-L1 on aAPCs was verified by flow cytometry (not shown) and the presence of the luciferase-expressing gene was verified by monitoring luciferase activity following anti-CD3 antibody stimulation. Antibiotic selection, flow cytometry and reporter expression served as cell line authentication method. The cells were periodically tested and found negative for Mycoplasma contamination using PCR-based method. Cytotoxicity assay[2] 5 000 ECs were seeded on transparent 96-well plates and cultured for 48 h in the presence of increasing concentrations of the BMS compounds [BMS-1001] or DMSO as a control (the concentration of DMSO was kept constant in all samples). Following the treatment, a metabolic activity test was performed with the use of Biolog Redox Dye Mix MB, according to the manufacturer's instructions. Flow cytometry measurements[2] Binding of sPD-L1 (aa 18–134) to ECs was evaluated by flow cytometry. The His-tagged PD-L1 protein or its mutants were stained with NTA-Atto 647 N fluorescent dye for 2 h at 22°C, at 8:1 molar ratio (protein:dye). PD-L1-Atto was formulated in 150 μl PBS with the tested compounds or antibodies. The samples were incubated for 30 min at 4°C in the dark. Meanwhile, ECs were centrifuged, washed with PBS and suspended in fresh PBS at concentration of 1 × 106 cells per ml. 50 μl of ECs was added to each sample and incubated on ice for additional 60 min. The final concentrations of the components were: 25 μg/ml of PD-L1 (1.5 μM), 125 μg/ml the anti-PD-L1 antibodies and control antibodies and 1 μM of the BMS compounds [BMS-1001] . The samples were analyzed using the BD FACS Verse flow cytometer and BD FACSuite v1.0.6 software. Cytotoxicity Assay: Modified Jurkat T cells (PD-1 Effector Cells, ECs) were seeded in 96-well plates and treated with increasing concentrations of BMS-1001 or DMSO control for 48 hours. Metabolic activity was then measured using a redox dye mix to determine cell viability and calculate EC₅₀ values. BMS-1001 showed low toxicity with an EC₅₀ of 33.4 µM.[2] PD-1/PD-L1 Checkpoint Assay: Artificial antigen-presenting cells (aAPCs, CHO-K1 cells expressing TCR ligand and PD-L1) were seeded in white 96-well plates. Serial dilutions of BMS-1001 in DMSO were prepared in RPMI medium with 1% FBS, keeping DMSO concentration constant. PD-1 Effector Cells (ECs, Jurkat cells expressing PD-1 and an NFAT-driven luciferase reporter) were added to the aAPCs in the presence of the compound dilutions. After 6 hours of co-culture at 37°C, luminescence was measured using a luciferase assay reagent. Luminescence indicates TCR-mediated activation released from PD-1/PD-L1 inhibition.[2] PD-1/sPD-L1 Effector Assay: 96-well plates were coated overnight at 4°C with anti-CD3 antibody. Soluble PD-L1 (sPD-L1) was pre-incubated with BMS-1001 or DMSO in PBS containing penicillin/streptomycin. This mixture was added to the antibody-coated wells. PD-1 Effector Cells (ECs) were then added. After 24 hours of culture, luciferase activity was measured to assess T-cell activation. Final concentrations of BMS-1001 tested were 0.12, 0.3, 1.2, and 3 µM, corresponding to molar ratios of BMS-1001 to sPD-L1 of 1:5, 1:2, 2:1, and 5:1.[2] Flow Cytometry Binding Assay: His-tagged soluble PD-L1 was labeled with a fluorescent NTA-dye. The labeled PD-L1 was pre-incubated with BMS-1001, control antibodies, or DMSO in PBS. PD-1-expressing Jurkat cells (ECs) were then added to the mixture and incubated on ice. After incubation, cells were analyzed by flow cytometry to measure fluorescence intensity, indicating the level of sPD-L1 binding to cell surface PD-1.[2] |
| Animal Protocol |
Pharmacokinetic Study in Male Sprague–Dawley Rats[3] Male Sprague–Dawley rats (200–220 g) were used to study the pharmacokinetics of compound NP19 [an analog of BMS-1001]. Diet was prohibited for 12 h before the experiment, but water was freely available. Blood samples (0.3 mL) were collected from the tail vein into heparinized 1.5 mL polythene tubes at 0.0833, 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 12, and 24 h after oral (10 mg/kg) or intravenous (1 mg/kg) administration of compound NP19. The compound was dissolved in 5% DMSO and 95% PEG-300 for intravenous administration or suspended in 0.5% sodium carboxymethyl cellulose (CMC-Na) for oral administration. The samples were immediately centrifuged at 3000g for 10 min. The plasma as-obtained (100 μL) was stored at −20 °C until analysis. PK parameters were determined from individual animal data using noncompartmental analysis in DAS (Drug and statistics) software. Instruments and analytical conditions for PK studies: A UPLC-MS/MS system with ACQUITY I-Class UPLC and a XEVO TQD triple quadrupole mass spectrometer (Waters Corp., Milford, MA, USA), equipped with an electrospray ionization (ESI) interface, was used to analyze the blood samples. The UPLC system was comprised of a Binary Solvent Manager (BSM) and a Sample Manager with Flow-Through Needle (SM-FTN). Masslynx 4.1 software (Waters Corp.) was used for data acquisition and instrument control. Multiple reaction monitoring (MRM) modes of m/z 555.35 → 181.03 for NP19 and m/z 237 → 194.1 for carbamazepine were utilized to conduct quantitative analysis. In Vivo Efficacy Study in Mouse B16F10 Melanoma Model[3] BALB/c mice, aged 6–8 weeks old, were used to study the inhibition effect of NP19 [an analog of BMS-1001]on subcutaneous transplanted model of melanoma cells. Murine B16F10 melanoma cells growing in a logarithmic growth phase were suspended in PBS at a density of 2 × 106 per mL. Each mouse was inoculated subcutaneously with 200 μL containing 4 × 105 cells. After tumors reached approximately 100 mm3 in volume, mice were divided into four groups randomly (n = 10) and treated with NP19 (25, 50, 100 mg/kg) and vehicle, respectively. The drugs were administered via intragastric gavage once a day for 15 days. The vehicle group was administered with 0.5% sodium carboxymethyl cellulose (CMC-Na). Animal activity and body weight were monitored during the entire experiment period to assess acute toxicity. Mice were sacrificed 16 days after the initiation of the treatment, and the tumor tissue and major organ (liver, spleen, thymus, and kidney) samples were collected. The harvested tumor tissue and organs (liver, kidney) were fixed in 4% paraformaldehyde, processed into paraffin routinely, stained with hematoxylin and eosin (H&E), and captured by microscope. Tumor growth inhibition value (TGI) was calculated using the formula: TGI(%) = [1 – Wt/Wv] × 100%, where Wt and Wv are the mean tumor weight of treatment group and vehicle control. In Vivo Efficacy Study in Mouse H22 Hepatoma Tumor Model[3] 6–8 weeks old male BALB/c mice were used. A total of 8 × 105 H22 cells were inoculated into the right flank of each mouse according to protocols of tumor transplant research. NP19 [an analog of BMS-1001]was dissolved in 5% DMSO, 40% PEG-200 and 55% saline solution to produce desired concentrations. Mice in control groups were injected intraperitoneally with 200 μL of vehicle solution only. Tumor volume was measured every 2 days with a traceable electronic digital caliper and calculated using the formula a × b2 × 0.5, where a and b represented the larger and smaller diameters, respectively. The mice were sacrificed after the treatments and tumors were excised and weighed. |
| Toxicity/Toxicokinetics |
BMS-1001 showed low unspecific toxicity in cell-based assays. The EC₅₀ for cytotoxicity in modified Jurkat T cells (PD-1 Effector Cells) after 48-hour treatment was 33.4 µM, as determined by a metabolic activity assay.[2] |
| References |
[1]. Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1. J Med Chem. 2017 Jul 13;60(13):5857-5867. [2]. Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget. 2017 Aug 7;8(42):72167-72181. [3]. Discovery of Novel Resorcinol Dibenzyl Ethers Targeting the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Interaction as Potential Anticancer Agents. J Med Chem. 2020 Aug 13;63(15):8338-8358. |
| Additional Infomation |
Antibodies targeting the PD-1/PD-L1 immune checkpoint achieved spectacular success in anticancer therapy in the recent years. In contrast, no small molecules with cellular activity have been reported so far. Here we provide evidence that small molecules are capable of alleviating the PD-1/PD-L1 immune checkpoint-mediated exhaustion of Jurkat T-lymphocytes. The two optimized small-molecule inhibitors of the PD-1/PD-L1 interaction, BMS-1001 and BMS-1166, developed by Bristol-Myers Squibb, bind to human PD-L1 and block its interaction with PD-1, when tested on isolated proteins. The compounds present low toxicity towards tested cell lines and block the interaction of soluble PD-L1 with the cell surface-expressed PD-1. As a result, BMS-1001 and BMS-1166 alleviate the inhibitory effect of the soluble PD-L1 on the T-cell receptor-mediated activation of T-lymphocytes. Moreover, the compounds were effective in attenuating the inhibitory effect of the cell surface-associated PD-L1. We also determined the X-ray structures of the complexes of BMS-1001 and BMS-1166 with PD-L1, which revealed features that may be responsible for increased potency of the compounds compared to their predecessors. Further development may lead to the design of an anticancer therapy based on the orally delivered immune checkpoint inhibition.[1] Blockade of the PD-1/PD-L1 immune checkpoint pathway with monoclonal antibodies has provided significant advances in cancer treatment. The antibody-based immunotherapies carry a number of disadvantages such as the high cost of the antibodies, their limited half-life, and immunogenicity. Development of small-molecule PD-1/PD-L1 inhibitors that could overcome these drawbacks is slow because of the incomplete structural information for this pathway. The first chemical PD-1/PD-L1 inhibitors have been recently disclosed by Bristol-Myers Squibb. Here we present NMR and X-ray characterization for the two classes of these inhibitors. The X-ray structures of the PD-L1/inhibitor complexes reveal one inhibitor molecule located at the center of the PD-L1 homodimer, filling a deep hydrophobic channel-like pocket between two PD-L1 molecules. Derivatives of (2-methyl-3-biphenylyl)methanol exhibit the structures capped on one side of the channel, whereas the compounds based on [3-(2,3-dihydro-1,4-benzodioxin-6-yl)-2-methylphenyl]methanol induce an enlarged interaction interface that results in the open "face-back" tunnel through the PD-L1 dimer.[2] BMS-1001 is a small-molecule inhibitor of the PD-1/PD-L1 immune checkpoint, developed by Bristol-Myers Squibb. It belongs to a class of compounds designed to block the interaction between PD-1 and PD-L1, thereby restoring T-cell function.[2] The crystal structure of BMS-1001 in complex with the Ig-like V-type domain of human PD-L1 was solved at 2.01 Å resolution. The compound binds in a tunnel at the interface of a PD-L1 dimer, inducing conformational changes, including rotation of the Tyr56 side chain. The (2R)-2-amino-3-hydroxypropanoic acid moiety forms hydrogen bonds with Asp122, and the 3-cyanobenzyl substituent makes hydrophobic contacts with Tyr123 and Arg125.[2] BMS-1001 induces dimerization of PD-L1 in solution, a mechanism proposed to contribute to its inhibitory action by occupying the PD-1 binding sites on both protomers.[2] The compound's activity in restoring T-cell activation, though demonstrated, is less potent than that of therapeutic anti-PD-1/PD-L1 antibodies.[2] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~12.5 mg/mL (~19.81 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 1.25 mg/mL (1.98 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 12.5 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 1.25 mg/mL (1.98 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 12.5 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.5845 mL | 7.9225 mL | 15.8451 mL | |
| 5 mM | 0.3169 mL | 1.5845 mL | 3.1690 mL | |
| 10 mM | 0.1585 mL | 0.7923 mL | 1.5845 mL |