BMH-21 (BMH 21; BMH21) is a novel and potent DNA intercalator with potential antineoplastic activity. It acts by binding to ribosomal DNA and inhibiting RNA polymerase I (Pol I) transcription. BMH-21 intercalated into dsDNA and exhibited a preference for binding to DNA sequences rich in GCs. With its broad range of cytotoxic effects on human cancer cell lines and its p53-independent action, BMH-21 is frequently used as a mediator of various cytotoxic drugs. By attaching to ribosomal (r) DNA, BMH-21 blocked RNA polymerase I (Pol I) transcription and degraded RPA194, the major catalytic subunit of Pol I.
Physicochemical Properties
| Molecular Formula | C21H20N4O2 | |
| Molecular Weight | 360.41 | |
| Exact Mass | 360.158 | |
| Elemental Analysis | C, 69.59; H, 6.12; N, 15.46; O, 8.83 | |
| CAS # | 896705-16-1 | |
| Related CAS # |
|
|
| PubChem CID | 3508054 | |
| Appearance | Yellow to orange solid powder | |
| Density | 1.3±0.1 g/cm3 | |
| Index of Refraction | 1.668 | |
| LogP | 1.57 | |
| Hydrogen Bond Donor Count | 1 | |
| Hydrogen Bond Acceptor Count | 4 | |
| Rotatable Bond Count | 4 | |
| Heavy Atom Count | 27 | |
| Complexity | 709 | |
| Defined Atom Stereocenter Count | 0 | |
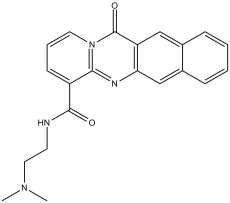
| SMILES | O=C(C1C2N(C(C3C(N=2)=CC2C(=CC=CC=2)C=3)=O)C=CC=1)NCCN(C)C |
|
| InChi Key | BXYDVWIAGDJBEC-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C21H20N4O2/c1-24(2)11-9-22-20(26)16-8-5-10-25-19(16)23-18-13-15-7-4-3-6-14(15)12-17(18)21(25)27/h3-8,10,12-13H,9,11H2,1-2H3,(H,22,26) | |
| Chemical Name | N-[2-(dimethylamino)ethyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | DNA | |||||||||||||||||||||||||
| ln Vitro |
BMH-21 inhibits Pol I transcription by binding to ribosomal DNA[1]. BMH-21 suppresses RNA polymerase I without regard to the DNA damage response[1]. BMH-21 prefers to bind to DNA sequences that are rich in GCs and intercalates into double strand (ds) DNA[1]. BMH-21 (1 μM; 3 hours) induces nucleolar stress and RPA194 degradation in a way that is independent of DNA damage[1]. Researchers have recently described a planar heterocyclic small molecule DNA intercalator, BMH-21, that binds ribosomal DNA and inhibits RNA polymerase I (Pol I) transcription. Despite DNA intercalation, BMH-21 does not cause phosphorylation of H2AX, a key biomarker activated in DNA damage stress. Here Researchers assessed whether BMH-21 activity towards expression and localization of Pol I marker proteins depends on DNA damage signaling and repair pathways. Researchers show that BMH-21 effects on the nucleolar stress response were independent of major DNA damage associated PI3-kinase pathways, ATM, ATR and DNA-PKcs. However, testing a series of BMH-21 derivatives with alterations in its N,N-dimethylaminocarboxamide arm showed that several derivatives had acquired the property to activate ATM- and DNA-PKcs -dependent damage sensing and repair pathways while their ability to cause nucleolar stress and affect cell viability was greatly reduced. The data show that BMH-21 is a chemically unique DNA intercalator that has high bioactivity towards Pol I inhibition without activation or dependence of DNA damage stress. The findings also show that interference with DNA and DNA metabolic processes can be exploited therapeutically without causing DNA damage.[1] Researchers show that the compound, BMH-21, has wide and potent antitumorigenic activity across NCI60 cancer cell lines. BMH-21 binds GC-rich sequences, which are present at a high frequency in ribosomal DNA genes, and potently and rapidly represses RNA polymerase I (Pol I) transcription. Strikingly, Researchers find that BMH-21 causes proteasome-dependent destruction of RPA194, the large catalytic subunit protein of Pol I holocomplex, and this correlates with cancer cell killing. Our results show that Pol I activity is under proteasome-mediated control, which reveals an unexpected therapeutic opportunity.[2] |
|||||||||||||||||||||||||
| ln Vivo |
BMH-21 (50 mg/kg; i.p.; daily; for 6 days) inhibits the growth of HCT116 colon cancer tumors in vivo[2]. BMH-21 also demonstrated potent antitumor activity at 25 mg/kg and 50 mg/kg in a human melanoma xenograft model and reduced Ki67 proliferation index (Figure 1G). Similarly, BMH-21 significantly inhibited HCT116 colon cancer tumor growth (Figure 1H). The tumor control ratios, calculated as change of median tumor weight over the course of treatment in the treatment group as compared to the control group were 21% and 30% for the A375 and HCT116 xenografts, respectively. BMH-21 did not cause changes in the weight of the mice or organ histology indicating that it is well tolerated. [2] |
|||||||||||||||||||||||||
| Enzyme Assay |
FID Assay [2] FID assay was conducted as in Tse and Boger, 2005. Deoxyoligonucleotide DNA hairpins containing 8-bp stem region with random DNA sequences or human rDNA sequences were purchased from .... EtBr (6 μM) was added to each well in sodium acetate buffer (35 mM NaOAc, 162.8 mM NaCl, 1.75 mM EDTA, pH 5.0), followed by hairpin DNA (1.5 μM), and 2 μM BMH-21 in sodium acetate buffer containing 2% DMSO. For each experiment the ratio of EtBr to hairpin DNA was adjusted to 1 molar equivalent EtBr per 2 DNA base pairs. The 0% control wells contained neither hairpin DNA nor BMH-21, while 100% control wells contained hairpin DNA but no BMH-21. All assays were conducted in triplicate. rRNA Synthesis Assays [2] Cells were labeled with 1 mM 5-FUrd (Sigma) using hypotonic shift and fixed with ice-cold methanol and acetone. Cells were blocked in 3% BSA and FUrd was detected using anti-5-BrdU antibody. DNA was stained with DAPI. Alternatively cells were labeled with ethynyl-uridine (EU) and processed according to the Click-IT protocol. The analysis of de novo synthesized rRNA was according to Pestov et al., 2008. Cells were labeled with [5, 6-3H]-uridine at 2–3 μCi/ml. Total RNA was isolated using NucleoSpin RNAII Total RNA isolation kit. 3–5 μg of total RNA was separated on a 0.8% agarose-formaldehyde gel and total 18S rRNA was detected for loading control. RNA was transferred to Hybond-N+ filter and crosslinked. The membrane was sprayed using Enhancer and exposed to film. In vitro Pol I Transcription Assay [2] In vitro Pol I transcription assay was conducted as in Mayer et al., 2004. The reaction mixtures contained 500 ng of template DNA containing Pol I promoter at −347 to +385 (pHrP2/EcoRI), HeLa nuclear extract, 10 mM HEPES-KOH, (pH 8.0), 0.1 mM EDTA, 10 mM MgCl2, 300 mM KCl, 10 mM creatine phosphate, 12.5% (v/v) glycerol, 0.66 mM each of ATP, GTP, UTP and CTP. After incubation for 2 hr at 30°C, RNA was extracted, and the rRNA product was amplified using PCR primers for 5′ETS rRNA located at +2 (forward),+82 (reverse), analyzed in 8% polyacrylamide gel and stained using SYBR Gold. In vitro Degradation Assay [2] In vitro degradation of RPA194 was as in Williamson et al., 2009 with slight modifications. Cells were treated with BMH-21 for 3 hr and extracts were prepared in SB-buffer (20 mM HEPES pH 7.5, 1.5 mM MgCl2, 5 mM KCl, 1 mM DTT, protease inhibitors), 15 mM creatine phosphate, 2 mM ATP). Reactions were carried out in a mix containing human recombinant E1 (32 ng/ml), UbcH5c (50 ng/ml), UbcH10 (10 ng/ml), ubiquitin (50 ng/ml), Ub-aldehyde and supplemented with 5 mM ATP. Reactions were initiated by addition of in vitro translated pRPA194-HA and incubated for 90–120 min at 30°C. MG132 was used at 85 μM when indicated. |
|||||||||||||||||||||||||
| Cell Assay |
The cells are kept in a humidified environment with 5% CO2 at 37 °C. The culture medium used for U2OS osteosarcoma cells contains 15% fetal bovine serum in addition to DMEM. In triplicate, cells are plated in 96-well plates at a density of 10,000 cells per well, and the compounds are then incubated for 48 hours. Using the WST-1 cell proliferation reagent, viability is assessed.
Viability assay[1] Cells were plated in 96-well plates at a density of 10,000 cells/well and incubated for 48 hours followed by viability measurement using the WST-1 cell proliferation reagent according to manufacturer's protocol. Immunofluorescence and image analysis[1] Cells grown on coverslips were fixed in 3.5% paraformaldehyde, permeabilized with 0.5% NP-40 and blocked in 3% BSA.The following primary antibodies were used: UBF (H-300), NCL (4E2), RPA194 (C-1), phospho-ATM, γH2AX, phospho-KAP1, phospho-DNA-PKcs. Secondary Alexa488 and Alexa594-cojugated anti-mouse and anti-rabbit antibodies were from Invitrogen. DNA was stained with DAPI. Images were captured using Axioplan2 fluorescence microscope (Zeiss) equipped with AxioCam HRc CCD-camera and AxioVision 4.5 software using EC Plan-Neofluar 20x/0.5 and 40x/0.75 objectives (Zeiss). Image analysis was conducted using FrIDA designed for the analysis of RGB color image datasets. Hue saturation and brightness ranges for green and red fluorescence channel and DNA (blue) were defined for each image set. Image intensities were determined as the fraction of positive cells divided total nuclear area as defined by DNA staining. An average of 100 cells was quantified from two fields for each sample. Immunoblotting[1] Cells were lysed in 0.5% NP-40 buffer (25 mM Tris-HCl, pH 8.0, 120 mM NaCl, 0.5% NP-40, 4 mM NaF, 100 μM Na3VO4, 100 KIU/ml aprotinin, 10 μg/ml leupeptin) or RIPA lysis buffer. Proteins were separated on SDS-PAGE, blotted, probed for respective proteins and detected using ECL). The primary antibodies used for detection were NCL, RPA194. HRP-conjugated secondary antibodies and were from DAKO or Santa Cruz Biotechnology, HRP-conjugated streptavidin was from DAKO. Chromatin Immunoprecipitation[2] A375 cells (1 × 107) were fixed with 1% formaldehyde, lysed, and chromatin was extracted essentially as in Denissov et al., 2011. All buffers were supplemented with 1x protease inhibitor cocktail. Chromatin was sheared to 500–1000 bp. Each IP was conducted using 100 μg DNA, 5 μg antibody, and collected using secondary antibody-coupled Dynabeads. DNA was purified with ChIP DNA Clean and Concentrator kit, and eluted to 100 μl. qPCR was conducted using 2 μl of elute in 10 μl reaction, using a final concentration of primers at 200 nM on ABI Prism7900. Primer sequences are listed in Supplemental Experimental Procedures. RNA Interference[2] U2OS cells were transfected with control siRNA duplex or specific siRNAs (10 nM) using Lipofectamine RNAiMAX and incubated for 48 hr. The following siRNAs were used: POLR1A si403, si405 and control siRNA. |
|||||||||||||||||||||||||
| Animal Protocol |
6-week old athymic NCr nu/nu mice, with HCT116 colorectal carcinoma xenograft 50 mg/kg Intraperitoneal injection, daily, for 6 days Mouse Xenograft Studies [2] Pharmacokinetic and toxicity studies are detailed in Supplemental Experimental Procedures. For tumor xenograft experiments, A375 (3×106) or HCT116 (2×106) cells were injected in 0.1 ml PBS to the flanks of 6-week old athymic NCr nu/nu mice and the tumors were allowed to establish until they reached 90–120 mm3. Mice were randomized to respective treatment groups (mock, BMH-21 25 mg/kg and BMH-21 50 mg/kg). Mock treatments consisted of phosphate-citrate buffer (pH 6.0). Treatments were administered i.p. on a cycle 6 days on, one day off (A375) or daily (HCT116). Tumor weight (TW) was calculated by measuring the tumor diameter with a caliper and using the formula: TW =( |
|