BI-882370 (BI 882370), structurally similar to vermurafenib, is a novel, highly potent and selective BRAF inhibitor with anticancer activity. In the DFG-out (inactive) conformation of the BRAF kinase, it binds to the ATP site of the RAF kinase. Given that commercial BRAF inhibitors bind to the DFG-in conformation, its binding to the DFG-out conformation is special. With IC50 values of 0.4, 0.8, and 0.6 nM, respectively, BI-882370 inhibits the oncogenic BRAFV600E-mutant, WT BRAF, and CRAF kinases. SRC family kinases are also inhibited by BI-882370. With 100× higher potency (1-10 nmol/L) than vemurafenib, BI 882370 prevents the proliferation of human BRAF-mutant melanoma cells.
Physicochemical Properties
| Molecular Formula | C28H33F2N7O2S |
| Molecular Weight | 569.669131040573 |
| Exact Mass | 569.238 |
| Elemental Analysis | C, 59.03; H, 5.84; F, 6.67; N, 17.21; O, 5.62; S, 5.63 |
| CAS # | 1392429-79-6 |
| Related CAS # | 1392429-79-6 |
| PubChem CID | 60152613 |
| Appearance | Off-white to gray solid powder |
| Density | 1.4±0.1 g/cm3 |
| Boiling Point | 666.8±65.0 °C at 760 mmHg |
| Flash Point | 357.0±34.3 °C |
| Vapour Pressure | 0.0±2.0 mmHg at 25°C |
| Index of Refraction | 1.660 |
| LogP | 2.59 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 10 |
| Rotatable Bond Count | 9 |
| Heavy Atom Count | 40 |
| Complexity | 913 |
| Defined Atom Stereocenter Count | 0 |
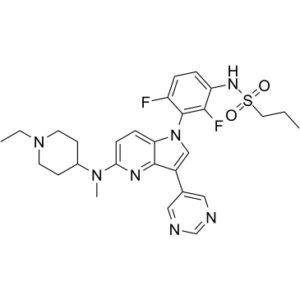
| SMILES | S(CCC)(NC1=CC=C(C(=C1F)N1C=C(C2=CN=CN=C2)C2=C1C=CC(=N2)N(C)C1CCN(CC)CC1)F)(=O)=O |
| InChi Key | AEJACXAFHXBVHF-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C28H33F2N7O2S/c1-4-14-40(38,39)34-23-7-6-22(29)28(26(23)30)37-17-21(19-15-31-18-32-16-19)27-24(37)8-9-25(33-27)35(3)20-10-12-36(5-2)13-11-20/h6-9,15-18,20,34H,4-5,10-14H2,1-3H3 |
| Chemical Name | N-[3-[5-[(1-ethylpiperidin-4-yl)-methylamino]-3-pyrimidin-5-ylpyrrolo[3,2-b]pyridin-1-yl]-2,4-difluorophenyl]propane-1-sulfonamide |
| Synonyms | BI882370; BI 882370; BI-882370 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Braf (Ki = 2 nM); JNK1 (IC50 = 45 nM); JNK2 (Ki = 4 nM); JNK2 (Ki = 160 nM); JNK3 (Ki = 52 nM) BI-882370 is a highly potent and selective RAF kinase inhibitor. It binds to the DFG-out (inactive) conformation of BRAF kinase. Biochemical inhibitory potencies (IC₅₀) are: BRAFV600E 0.4 nM, BRAFWT 0.8 nM, CRAF 0.6 nM. Dissociation constants (KD) determined by SPR are: BRAFV600E 4 nM, BRAFWT 6 nM, CRAF 3 nM. [1] |
| ln Vitro |
BI-882370 (0.9-6000 nM; 3 days) has an EC50 range of 1–10 nM and inhibits the proliferation of human melanoma and colorectal cancer cells that have the BRAF mutation[1]. BI 882370 (0.1-100 nM, 0.1-3000 nM; 2 hours) reduces the expression of p-MEK1/2, p-ERK1/2, and cyclin D1/D2 in BRAFV600E-mutant A375 cells; in WT BRO cells, it increases the phosphorylation of ERK1/2 and phosphorylates MEK1/2 (3–300 nM)[1]. In BRAFV600E-mutant A375 cells, BI 882370 (0.1-100 nM, 0.1-3000 nM; 24 hours) inhibits cyclin D1/D2 expression and increases Kip1/p27 expression at concentrations of 1 nM or higher. Expression of cyclins D1/D2 or Kip1/p27 is unaffected in WT BRO cells. BI-882370 inhibited proliferation of human BRAF-mutant melanoma cells (e.g., A375, SK-MEL-28, G-361) with EC₅₀ values in the range of 0.9 to 6 nM, which was approximately 100-fold more potent than vemurafenib. Wild-type BRAF cell lines (e.g., BRO, HCT-116) were not affected at concentrations up to 1,000 nM, with EC₅₀ values > 5,000 nM. [1] In A375 melanoma cells (BRAFV600E), treatment with BI-882370 for 2 hours resulted in a dose-dependent reduction of phospho-MEK1/2 and phospho-ERK1/2 levels in immunoblots, with complete suppression observed at 3 nM. After 24 hours of treatment, it suppressed cyclin D1/D2 expression and induced Kip1/p27 expression, indicating G1 phase cell cycle arrest. [1] In cell-based ELISA assays, BI-882370 inhibited ERK phosphorylation with EC₅₀ values of 0.5 nM in A375 cells and 0.7 nM in SK-MEL-28 cells. [1] BI-882370 did not induce significant RAF heterodimer (CRAF/BRAF or CRAF/ARAF) formation in BRAF wild-type, NRAS-mutant BRO melanoma cells at pharmacologically relevant concentrations, unlike the tool compound GDC-0879. However, it did induce phosphorylation of MEK and ERK in these cells at concentrations between 3 and 300 nM, without affecting cyclin D1/D2 or Kip1/p27 expression. [1] The effect of BI-882370 on pathway inhibition was long-lasting. After a 2-hour treatment and subsequent washout in A375 cells, suppression of ERK phosphorylation persisted, with EC₅₀ values still in the 10-20 nM range 24 hours post-washout. This contrasts with vemurafenib, where pathway activity returned to baseline within 1 hour after washout. [1] |
| ln Vivo |
BI-882370 (deliver orally; 25 mg/kg, 50 mg/kg; twice daily; 2 weeks) is more effective than Vemurafenib, Dabrafenib, or Trametinib in a number of mouse models of colorectal and BRAF-mutant melanoma[1]. BI-882370 (deliver orally; 25 mg/kg; twice daily; 40 days) develops resistance within three weeks, but trametinib-assisted second-line therapy for five weeks does not reveal resistance[1]. BI-882370 (deliver orally; 60 mg/kg; once daily; 2 weeks) shows no toxicity in rats when measured by clinical chemistry, hematology, pathology, and toxicogenomics[1]. In mouse xenograft models of BRAFV600E melanoma (A375), oral administration of BI-882370 at 25 mg/kg twice daily (bid) induced complete or partial regression of all tumors, showing superior efficacy compared to vemurafenib (120 mg/kg once daily, qd) and dabrafenib (60 mg/kg qd). [1] In the G-361 melanoma model (heterozygous BRAFV600V/E), BI-882370 (12.5 or 25 mg/kg bid) induced tumor regression, whereas vemurafenib only led to initial stabilization followed by regrowth. [1] In a model of acquired resistance, A375 tumors that progressed after initial vemurafenib treatment were re-treated. BI-882370 (25 mg/kg bid) as a single agent induced tumor regression, but resistance developed within 2-3 weeks. The combination of BI-882370 (25 mg/kg bid) with the MEK inhibitor trametinib (0.25 mg/kg bid) resulted in more pronounced regression without regrowth during a 5-week second-line treatment period. [1] In BRAFV600E colorectal cancer models, BI-882370 (25 mg/kg bid) induced complete or partial regression in COLO 205 tumors and showed significant tumor growth inhibition (TGI) in the less sensitive HT-29 model. [1] Combination therapy with BI-882370 and the EGFR inhibitor cetuximab or the ErbB inhibitor afatinib, or with trametinib, synergistically improved efficacy in the HT-29 colorectal cancer model. [1] Immunohistochemical analysis of tumor samples from treated mice showed strong suppression of phospho-ERK signal. [1] |
| Enzyme Assay |
A RAF-MEK-ERK cascade assay was used to determine RAF kinase inhibition. The activity of ERK on a fluorogenic substrate was measured using Z-LYTE technology. Briefly, the assay components (RAF, MEK, ERK kinases and substrate) were combined in buffer, and compounds were added in a dose-response manner. After incubation, the fluorescence signal was measured, and IC₅₀ values were calculated by non-linear regression. [1] Surface plasmon resonance (SPR) was used to determine the binding affinity (KD) of compounds to purified BRAF and CRAF kinase domains. The protein was immobilized on a sensor chip, and compounds at various concentrations were flowed over the surface. The association and dissociation rates were monitored in real-time, and equilibrium dissociation constants were calculated. [1] |
| Cell Assay |
For proliferation assays, cells were seeded into 96-well plates. The next day, serially diluted compounds were added (final DMSO concentration 1%). After 3 days of treatment, the antiproliferative effect was determined. For most cell lines, metabolic activity of remaining cells was measured using alamarBlue reagent. Fluorescence was read, and EC₅₀ values were calculated by non-linear regression. For HT-29 and COLO 205 cells, a 3H-thymidine incorporation assay was used instead. After 3 days of compound treatment, 3H-thymidine was added for 16 hours. Cells were then lysed, DNA was harvested onto filter plates, and radioactivity was measured using a microplate scintillation counter to determine EC₅₀. [1] For phospho-ERK cell-based ELISA, A375 or SK-MEL-28 cells were plated, treated with compounds for 2 hours, and then fixed. Cells were permeabilized, blocked, and incubated overnight with an anti-phospho-ERK primary antibody. After washing, an HRP-conjugated secondary antibody was added, followed by TMB substrate. The reaction was stopped, and absorbance was measured at 450 nm to determine EC₅₀ values. [1] For immunoblotting, cells were treated with compounds for 2 or 24 hours, lysed, and proteins were separated by SDS-PAGE. After transfer to a membrane, blots were probed with antibodies against phospho-MEK, MEK, phospho-ERK, ERK, cyclin D1/2, Kip1/p27, and α-tubulin (loading control), followed by ECL detection. [1] For co-immunoprecipitation to assess RAF dimerization, cells were treated for 1 hour, lysed, and CRAF was immunoprecipitated using a specific antibody. The immunoprecipitates were then analyzed by Western blotting using antibodies against ARAF, BRAF, and CRAF. [1] |
| Animal Protocol |
Human melanoma xenografts in nude mice with BRAF-mutant melanomas and colorectal carcinomas cells (A375, COLO 205; G-361, HT-29 cells)[1] 25 mg/kg; 50 mg/kg Deliver orally; 25 mg/kg, 50 mg/kg; twice daily; 2 weeks For efficacy studies in mouse xenograft models, female immunodeficient mice were subcutaneously injected with tumor cells (e.g., A375, COLO 205). When tumors were well-established (approx. 50-100 mm³), mice were randomized into treatment groups. [1] BI-882370 was formulated as a solution in 0.5% Natrosol (hydroxyethylcellulose), adjusted to pH 3 with citric acid. [1] Compounds were administered orally (intragastrically) at a volume of 10 mL/kg body weight. BI-882370 was typically dosed twice daily (bid) at 6.25, 12.5, or 25 mg/kg. Treatment duration varied from 2 to 6 weeks depending on the experiment. [1] Tumor diameters were measured 3 times per week, and volumes were calculated. Body weight was monitored as a measure of tolerability. [1] For pharmacodynamic and pharmacokinetic analyses, blood samples and tumors were collected at specified time points after the last dose for drug concentration measurement or for immunohistochemical analysis. [1] |
| ADME/Pharmacokinetics |
In tumor-bearing mice treated with BI-882370 at 25 mg/kg twice daily for 2-3 weeks, the drug exposure (AUC0-24h) at the end of the treatment period ranged from 12,000 to 25,000 nmol·h/L. [1] After a single oral dose of BI-882370 (50 mg/kg) in mice, drug concentrations in tumor tissue were higher than in plasma at all time points investigated, indicating favorable tissue distribution. [1] In rats, a dose of 60 mg/kg once daily for 2 weeks resulted in an exposure (AUC) of 43,000 nmol·h/L. [1] |
| Toxicity/Toxicokinetics |
BI-882370 was well tolerated in mice at doses up to 50 mg/kg twice daily for several weeks, with no significant body weight loss or clinical signs of toxicity observed. A maximum tolerated dose (MTD) was not defined in these studies. [1] Histopathological examination of multiple organs (skin, heart, lung, bladder, stomach, liver, kidney) from tumor-bearing mice treated with BI-882370 at highly efficacious doses (25 mg/kg bid) for 2 weeks revealed no evidence of drug-induced hyperplasia or other pathological alterations. [1] In an exploratory 2-week toxicology study in male rats, daily oral doses of BI-882370 up to 60 mg/kg (resulting in AUC > 43,000 nmol·h/L) did not cause any adverse clinical observations or relevant findings in clinical chemistry, hematology, histopathology, or toxicogenomic analysis of liver and skin. [1] |
| References |
[1]. A Novel RAF Kinase Inhibitor with DFG-Out-Binding Mode: High Efficacy in BRAF-Mutant Tumor Xenograft Models in the Absence of Normal Tissue Hyperproliferation. Mol Cancer Ther. 2016 Mar;15(3):354-65. |
| Additional Infomation |
pan-RAF Inhibitor XP-102 is an orally bioavailable, second-generation inhibitor of all members of the serine/threonine protein kinase Raf family, including A-Raf, B-Raf and C-Raf protein kinases, with potential antineoplastic activity. Upon administration, pan-RAF kinase inhibitor XP-102 specifically binds to the ATP binding site of the Raf kinase positioned in the inactive DFG-out conformation, and inhibits the activity of Raf, including B-Raf mutated forms such as the B-Raf V600E mutation. This prevents the activation of Raf-mediated signal transduction pathways, which may inhibit tumor cell growth. Raf protein kinases play a key role in the RAF/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling pathway, which is often dysregulated in human cancers and plays a key role in tumor cell proliferation and survival. The valine to glutamic acid substitution at residue 600 accounts for the majority of BRAF gene mutations. The oncogenic product, BRAF(V600E) kinase, exhibits elevated activity that over-activates the MAPK signaling pathway. BI-882370 is a DFG-out (Type II) kinase inhibitor, binding to the inactive conformation of BRAF, in contrast to first-generation inhibitors like dabrafenib and vemurafenib which are DFG-in (Type I) binders. [1] Its binding mode stabilizes the DFG-out conformation via specific hydrogen bonds and a T-stacking interaction with Phe595, contributing to its high potency. [1] A key feature of BI-882370 is its lack of induction of RAF heterodimer formation in BRAF wild-type cells at pharmacologically relevant concentrations, which is associated with the paradoxical activation of the MAPK pathway and hyperproliferative side effects (like skin lesions) seen with some other RAF inhibitors. [1] BI-882370 demonstrates a long duration of action in cells and in vivo, providing sustained pathway suppression. [1] It shows superior efficacy as a single agent compared to vemurafenib, dabrafenib, and trametinib in multiple BRAF-mutant xenograft models when dosed to achieve exposures comparable to those in patients. [1] BI-882370 in combination with trametinib showed promising activity in a model of acquired resistance to vemurafenib. [1] |
Solubility Data
| Solubility (In Vitro) |
DMSO : ~5 mg/mL (~8.78 mM) H2O : < 0.1 mg/mL |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.7554 mL | 8.7770 mL | 17.5540 mL | |
| 5 mM | 0.3511 mL | 1.7554 mL | 3.5108 mL | |
| 10 mM | 0.1755 mL | 0.8777 mL | 1.7554 mL |