Physicochemical Properties
| Molecular Formula | C20H15N3O2CL2 |
| Molecular Weight | 400.258 |
| Exact Mass | 445.131 |
| CAS # | 1505453-59-7 |
| PubChem CID | 72203613 |
| Appearance | White to off-white solid powder |
| LogP | 4.607 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 5 |
| Rotatable Bond Count | 4 |
| Heavy Atom Count | 32 |
| Complexity | 727 |
| Defined Atom Stereocenter Count | 1 |
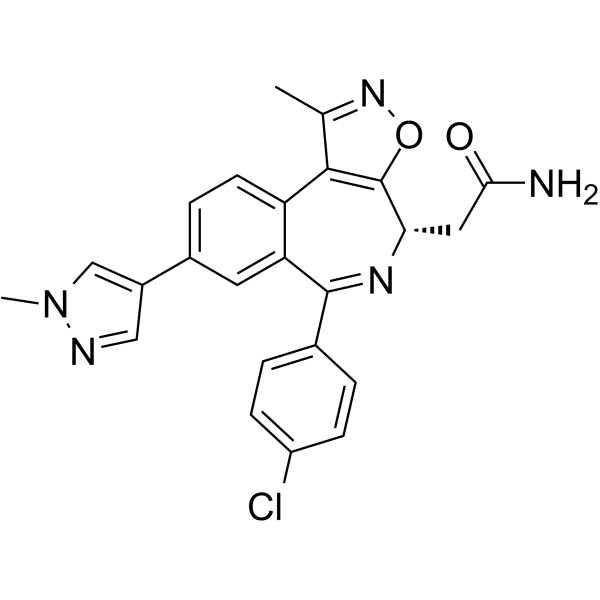
| SMILES | CC1=NOC2=C1C3=C(C=C(C=C3)C4=CN(N=C4)C)C(=N[C@H]2CC(=O)N)C5=CC=C(C=C5)Cl |
| InChi Key | QFLGNZXBWIQDLQ-FQEVSTJZSA-N |
| InChi Code | InChI=1S/C24H20ClN5O2/c1-13-22-18-8-5-15(16-11-27-30(2)12-16)9-19(18)23(14-3-6-17(25)7-4-14)28-20(10-21(26)31)24(22)32-29-13/h3-9,11-12,20H,10H2,1-2H3,(H2,26,31)/t20-/m0/s1 |
| Chemical Name | 2-[(4S)-6-(4-chlorophenyl)-1-methyl-8-(1-methylpyrazol-4-yl)-4H-[1,2]oxazolo[5,4-d][2]benzazepin-4-yl]acetamide |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| ln Vitro | Compound 38, BET bromodomain inhibitor 1 (31.25-125 nM; 24 hr) induces a more prominent G1-phase cell cycle arrest[1]. The highly effective BET bromodomain inhibitor 1 (31.25-500 nM; 6 or 24 hours) inhibits c-Myc expression in a dose-dependent manner and upregulates p21 levels[1]. BET bromodomain inhibitor 1 (31.25-125 nM; 6 hours) significantly lowers CDK6, BCL-2, and c-Myc expressions.[1]. Five cytochrome P450 enzymes (IC50>20 μM) are not inhibited by BET bromodomain inhibitor 1[1]. With an approximately 15000-fold selectivity for BRD4(1) over EP300 (IC50=3857 nM), BET bromodomain inhibitor 1 exhibits exceptional selectivity for the BET bromodomain family over other bromodomains[1]. With IC50 values of 2.4, 4.8, 17.6, and 15.1 nM, respectively, BET bromodomain inhibitor 1 significantly suppressed the growth of acute myeloid leukemia cell line MV4-11, acute leukemia cell lines Kasumi-1 and RS-4-11, and multiple myeloma cancer cell line MM1.S cells[1]. |
| ln Vivo | Compound 38, also known as BET bromodomain inhibitor 1 (PO; 6.25, 12.5 mg/kg; daily; for 28 days), has more potent anticancer effects and totally stops tumor development at 12.5 mg/kg (tumor growth inhibition [TGI] of 99.7% [1]. For rats and mice, respectively, BET bromodomain inhibitor 1 (1 mg/kg; IV) had a T1/2 of 1.3 and 0.9 hours, a CL of 21.5 and 15.3 mL/min·kg, and a Vss of 1464 and 782 mL/kg[1]. Rats' T1/2 is 3.6 hours, Cmax is 159 ng/mL, and AUC is 884 ng·h/mL for BET bromodomain inhibitor 1 (3 mg/kg; PO)[1]. For mice, BET bromodomain inhibitor 1 (1.3 mg/kg; PO) shows an AUC of 1710 ng·h/mL, a Cmax of 399 ng/mL, and a T1/2 of 1.3 hours[1]. |
| Cell Assay |
Cell Cycle Analysis[1] Cell Types: MV-4-11 cells Tested Concentrations: 31.25, 62.5, 125 nM Incubation Duration: 24 hrs (hours) Experimental Results: Led to more pronounced G1-phase cell cycle arrest. Western Blot Analysis[1] Cell Types: MV-4-11 cells Tested Concentrations: 31.25, 62.5, 125, 250, 500 nM Incubation Duration: 6 or 24 hrs (hours) Experimental Results: Induced dose-dependent inhibition on c-Myc expression and upregulation of p21 levels. RT-PCR[1] Cell Types: MV-4-11 cells Tested Concentrations: 31.25, 62.5, 125 nM Incubation Duration: 6 hrs (hours) Experimental Results: Robustly decreased the expressions of c-Myc, BCL-2, and CDK6. |
| Animal Protocol |
Animal/Disease Models: An MV4-11 mouse xenograft model[1] Doses: 6.25, 12.5 mg/kg Route of Administration: PO; daily; for 28 days Experimental Results: demonstrated stronger antitumor activities and completely inhibited the growth of tumor with a tumor growth inhibition (TGI) of 99.7 % at 12.5 mg/kg. Animal/Disease Models: Male SD rats[1] Doses: 1 mg/kg (pharmacokinetic/PK Analysis) Route of Administration: IV Experimental Results: Had a T1/2 of 1.3 hrs (hours), a CL of 21.5 mL/min·kg, and a Vss of 1464 mL/kg. |
| References |
[1]. WO/2015/153871A2. |
Solubility Data
| Solubility (In Vitro) | DMSO : ≥ 100 mg/mL (~224.27 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.61 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.61 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (5.61 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4984 mL | 12.4919 mL | 24.9838 mL | |
| 5 mM | 0.4997 mL | 2.4984 mL | 4.9968 mL | |
| 10 mM | 0.2498 mL | 1.2492 mL | 2.4984 mL |