Balicatib (also known as AAE-581) is a novel, potent and selective cathepsin K inhibitor that, in cell-based enzyme occupancy assays, has 10-100 times the potency of cathepsin B, L, and S combined. Balicatib inhibited bone turnover at most sites, partially prevented changes in bone mass caused by ovariectomy, and had an unexpected stimulatory effect on periosteal bone formation because it inhibits the osteoclastic enzyme cathepsin K.
Physicochemical Properties
| Molecular Formula | C23H33N5O2 |
| Molecular Weight | 411.5404 |
| Exact Mass | 411.263 |
| Elemental Analysis | C, 67.12; H, 8.08; N, 17.02; O, 7.78 |
| CAS # | 354813-19-7 |
| Related CAS # | 354813-19-7 |
| PubChem CID | 10201696 |
| Appearance | white to off-white solid powder |
| Density | 1.2±0.1 g/cm3 |
| Boiling Point | 687.4±55.0 °C at 760 mmHg |
| Flash Point | 369.5±31.5 °C |
| Vapour Pressure | 0.0±2.1 mmHg at 25°C |
| Index of Refraction | 1.590 |
| LogP | 1.56 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 5 |
| Rotatable Bond Count | 7 |
| Heavy Atom Count | 30 |
| Complexity | 621 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | O=C(C1(C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H])N([H])C(C1C([H])=C([H])C(=C([H])C=1[H])N1C([H])([H])C([H])([H])N(C([H])([H])C([H])([H])C([H])([H])[H])C([H])([H])C1([H])[H])=O)N([H])C([H])([H])C#N |
| InChi Key | LLCRBOWRJOUJAE-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C23H33N5O2/c1-2-14-27-15-17-28(18-16-27)20-8-6-19(7-9-20)21(29)26-23(10-4-3-5-11-23)22(30)25-13-12-24/h6-9H,2-5,10-11,13-18H2,1H3,(H,25,30)(H,26,29) |
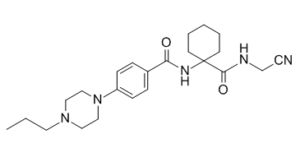
| Chemical Name | N-[1-(cyanomethylcarbamoyl)cyclohexyl]-4-(4-propylpiperazin-1-yl)benzamide |
| Synonyms | Balicatib; AAE-581; AAE 581; AAE581 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
cathepsin K (IC50 = 22 nM); cathepsin L (IC50 = 48 nM); Cathepsin B (IC50 = 61 nM); cathepsin S (IC50 = 2900 nM) Balicatib (0-10 µM) exhibits less than 1.5-fold accumulation of Type I collagen at concentrations up to 10 µM in human dermal fibroblasts[2]. Human cathepsin K (a cysteine protease), reversible inhibitor. [1] |
| ln Vitro |
Balicatib (0-10 µM) exhibits less than 1.5-fold accumulation of Type I collagen at concentrations up to 10 µM in human dermal fibroblasts[2]. In a functional bone resorption assay using rabbit osteoclasts cultured on bovine bone, Balicatib had a corrected bone resorption IC₅₀ of 22 nM (calculated based on the difference between rabbit and human Cat K potencies). This potency was 3- to 4-fold lower than that of odanacatib and L-873724. [2] In whole cell enzyme occupancy assays using human HepG2 cells (for Cat B and L occupancy) and Ramos cells (for Cat S occupancy), Balicatib showed IC₅₀ values of 61 nM (Cat B), 48 nM (Cat L), and 2900 nM (Cat S), indicating poor cellular selectivity compared to its potency on Cat K. [2] In a cellular model using primary human dermal fibroblasts (HDF) cultured in a 3-D collagen gel, treatment with 1-10 μM Balicatib for 3 days caused a 3- to 7-fold accumulation of intracellular Type I collagen, as quantified by flow cytometry using an anti-type I collagen antibody. This effect was attributed to its inhibition of multiple cathepsins due to its lysosomotropic properties. [2] In a functional bone resorption assay using rabbit osteoclasts cultured on bovine bone, Balicatib had a corrected bone resorption IC₅₀ of 22 nM (calculated based on the difference between rabbit and human Cat K potencies). This potency was 3- to 4-fold lower than that of odanacatib and L-873724. [2] In whole cell enzyme occupancy assays using human HepG2 cells (for Cat B and L occupancy) and Ramos cells (for Cat S occupancy), Balicatib showed IC₅₀ values of 61 nM (Cat B), 48 nM (Cat L), and 2900 nM (Cat S), indicating poor cellular selectivity compared to its potency on Cat K. [2] In a cellular model using primary human dermal fibroblasts (HDF) cultured in a 3-D collagen gel, treatment with 1-10 μM Balicatib for 3 days caused a 3- to 7-fold accumulation of intracellular Type I collagen, as quantified by flow cytometry using an anti-type I collagen antibody. This effect was attributed to its inhibition of multiple cathepsins due to its lysosomotropic properties. [2] |
| ln Vivo |
Balicatib (0, 3, 10, 50 mg/kg; Oral gavage; twice daily for 18 months) inhibited bone turnover at most sites, slightly prevented changes in bone mass brought on by ovariectomy, and stimulated the formation of periosteal bone in cynomolgus monkeys[1]. In ovariectomized cynomolgus monkeys (a postmenopausal osteoporosis model), treatment with Balicatib for 18 months partially prevented ovariectomy-induced bone mineral density (BMD) loss in the lumbar spine and increased BMD in the femur above levels seen in both ovariectomized and sham-ovariectomized controls. [1] Balicatib treatment (3, 10, 30 mg/kg/day) significantly inhibited bone turnover at most skeletal sites, as shown by reduced bone formation rates (BFR/BS) in cancellous bone of the vertebra and femur neck, and in endocortical and osteomal bone envelopes. [1] Unexpectedly, Balicatib treatment significantly stimulated periosteal bone formation. Periosteal bone formation rate (Ps.BFR/BS) was significantly increased at the lumbar vertebra cortex (high dose), femur neck cortex (medium and high doses), and femur midshaft cortex (all doses) compared to ovariectomized controls. [1] In ovariectomized cynomolgus monkeys (a postmenopausal osteoporosis model), treatment with Balicatib for 18 months partially prevented ovariectomy-induced bone mineral density (BMD) loss in the lumbar spine and increased BMD in the femur above levels seen in both ovariectomized and sham-ovariectomized controls. [1] Balicatib treatment (3, 10, 30 mg/kg/day) significantly inhibited bone turnover at most skeletal sites, as shown by reduced bone formation rates (BFR/BS) in cancellous bone of the vertebra and femur neck, and in endocortical and osteomal bone envelopes. [1] Unexpectedly, Balicatib treatment significantly stimulated periosteal bone formation. Periosteal bone formation rate (Ps.BFR/BS) was significantly increased at the lumbar vertebra cortex (high dose), femur neck cortex (medium and high doses), and femur midshaft cortex (all doses) compared to ovariectomized controls. [1] |
| Enzyme Assay |
The in vitro activity (IC₅₀) of Balicatib against human Cat K and off-target cathepsins (B, L, S) was determined using purified enzyme assays. The assay conditions are referenced from a previous publication. IC₅₀ values represent an average of at least three titrations, with standard deviations typically within 35% of the IC₅₀ values. [2] The in vitro activity (IC₅₀) of Balicatib against human Cat K and off-target cathepsins (B, L, S) was determined using purified enzyme assays. The assay conditions are referenced from a previous publication. IC₅₀ values represent an average of at least three titrations, with standard deviations typically within 35% of the IC₅₀ values. [2] |
| Cell Assay |
The potency of Balicatib in a functional bone resorption assay was evaluated using rabbit osteoclasts cultured on bovine bone slices. The assay measures the inhibition of bone resorption pit formation by osteoclasts. [2] Whole cell enzyme occupancy assays were performed to evaluate selectivity in a more physiologically relevant context. Human HepG2 cells were used to assess occupancy of Cat B and Cat L, and human Ramos B cells were used to assess occupancy of Cat S. Cells were treated with the compound, lysed, and enzyme occupancy was measured using activity-based probes. [2] The effect on intracellular collagen accumulation was assessed using primary human dermal fibroblasts (HDF). Fibroblasts were cultured within a three-dimensional collagen gel. Inhibitors were added for the final 3 days of culture. Subsequently, cells were extracted from the collagen matrix using collagenase treatment, then fixed, permeabilized, and stained intracellularly for Type I collagen using a specific monoclonal primary antibody and a FITC-labeled secondary antibody. The mean fluorescence intensity per cell, corresponding to intracellular collagen levels, was quantified by flow cytometry. [2] The potency of Balicatib in a functional bone resorption assay was evaluated using rabbit osteoclasts cultured on bovine bone slices. The assay measures the inhibition of bone resorption pit formation by osteoclasts. [2] Whole cell enzyme occupancy assays were performed to evaluate selectivity in a more physiologically relevant context. Human HepG2 cells were used to assess occupancy of Cat B and Cat L, and human Ramos B cells were used to assess occupancy of Cat S. Cells were treated with the compound, lysed, and enzyme occupancy was measured using activity-based probes. [2] The effect on intracellular collagen accumulation was assessed using primary human dermal fibroblasts (HDF). Fibroblasts were cultured within a three-dimensional collagen gel. Inhibitors were added for the final 3 days of culture. Subsequently, cells were extracted from the collagen matrix using collagenase treatment, then fixed, permeabilized, and stained intracellularly for Type I collagen using a specific monoclonal primary antibody and a FITC-labeled secondary antibody. The mean fluorescence intensity per cell, corresponding to intracellular collagen levels, was quantified by flow cytometry. [2] |
| Animal Protocol |
11-13 years, female cynomolgus monkeys (Macaca fascicularis)[1] 0, 3, 10, 50 mg/kg Oral gavage; twice daily for 18 months A total of 100 adult female cynomolgus monkeys (Macaca fascicularis) aged 11-13 years were used. Eighty animals underwent bilateral ovariectomy (Ovx), and 20 underwent sham surgery. Ovx animals were divided into four groups: vehicle control (Group O), and three Balicatib treatment groups receiving 3 (Low, L), 10 (Medium, M), or 50/30 (High, H) mg/kg/day. The high dose was reduced from 50 to 30 mg/kg/day approximately 1 month after initiation due to reduced food consumption. [1] Balicatib (AAE581 maleate) was dissolved in sterile water. Dose solutions were prepared weekly and refrigerated. The compound was administered orally twice daily via nasogastric intubation, starting the day after surgery, for 18 months. [1] Bone mineral density (BMD) of the lumbar spine (L2-4) and whole femur was measured by DXA at baseline and at 3, 6, 9, 12, 15, and 18 months under ketamine sedation. [1] Prior to necropsy at 18 months, bone formation was labeled by intravenous administration of calcein (4 mg/kg) on a schedule (label, 14-day interval, label, 7-day interval, necropsy). [1] At necropsy, the second lumbar vertebra and right femur were collected, fixed in 70% ethanol, and processed for static and dynamic bone histomorphometry to assess parameters such as mineralizing surface/bone surface (MS/BS), mineral apposition rate (MAR), and bone formation rate/bone surface (BFR/BS). [1] A total of 100 adult female cynomolgus monkeys (Macaca fascicularis) aged 11-13 years were used. Eighty animals underwent bilateral ovariectomy (Ovx), and 20 underwent sham surgery. Ovx animals were divided into four groups: vehicle control (Group O), and three Balicatib treatment groups receiving 3 (Low, L), 10 (Medium, M), or 50/30 (High, H) mg/kg/day. The high dose was reduced from 50 to 30 mg/kg/day approximately 1 month after initiation due to reduced food consumption. [1] Balicatib (AAE581 maleate) was dissolved in sterile water. Dose solutions were prepared weekly and refrigerated. The compound was administered orally twice daily via nasogastric intubation, starting the day after surgery, for 18 months. [1] Bone mineral density (BMD) of the lumbar spine (L2-4) and whole femur was measured by DXA at baseline and at 3, 6, 9, 12, 15, and 18 months under ketamine sedation. [1] Prior to necropsy at 18 months, bone formation was labeled by intravenous administration of calcein (4 mg/kg) on a schedule (label, 14-day interval, label, 7-day interval, necropsy). [1] At necropsy, the second lumbar vertebra and right femur were collected, fixed in 70% ethanol, and processed for static and dynamic bone histomorphometry to assess parameters such as mineralizing surface/bone surface (MS/BS), mineral apposition rate (MAR), and bone formation rate/bone surface (BFR/BS). [1] |
| ADME/Pharmacokinetics |
Balicatib is described as a lysosomotropic compound. Its basic and lipophilic nature leads to accumulation in lysosomes (pH 4-5), which increases its apparent potency against off-target cathepsins also located in lysosomes in cellular assays. [2] Balicatib is described as a lysosomotropic compound. Its basic and lipophilic nature leads to accumulation in lysosomes (pH 4-5), which increases its apparent potency against off-target cathepsins also located in lysosomes in cellular assays. [2] |
| Toxicity/Toxicokinetics |
Food consumption fell abruptly in the high-dose group (50 mg/kg/day) upon initiation of dosing, reaching a nadir at week 4, leading to a dose reduction to 30 mg/kg/day. [1] Body weight gain during the study was significantly lower in the high-dose group compared to ovariectomized controls. [1] One animal in the high-dose group died during month 16 following clinical signs of hind limb paralysis. Retrospectively, this animal had abnormalities including high serum osteocalcin and CTx levels since baseline and steady spinal bone loss. [1] Food consumption fell abruptly in the high-dose group (50 mg/kg/day) upon initiation of dosing, reaching a nadir at week 4, leading to a dose reduction to 30 mg/kg/day. [1] Body weight gain during the study was significantly lower in the high-dose group compared to ovariectomized controls. [1] One animal in the high-dose group died during month 16 following clinical signs of hind limb paralysis. Retrospectively, this animal had abnormalities including high serum osteocalcin and CTx levels since baseline and steady spinal bone loss. [1] |
| References |
[1]. Balicatib, a cathepsin K inhibitor, stimulates periosteal bone formation in monkeys. Osteoporos Int. 2012 Jan;23(1):339-49. [2]. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg Med Chem Lett. 2008 Feb 1;18(3):923-8. |
| Additional Infomation |
Balicatib has been used in trials studying the treatment of Osteoporosis and Knee Osteoarthritis. Balicatib is a cathepsin K inhibitor developed for the treatment of osteoporosis. Unlike most bone resorption inhibitors (e.g., bisphosphonates), Balicatib demonstrated a unique ability to stimulate periosteal bone formation in this long-term primate study, suggesting it may have anabolic properties in addition to its anti-resorptive effects. [1] The study suggests that the stimulatory effect on periosteal bone formation, combined with weak inhibition of endocortical bone formation in the femur, may explain why Balicatib increased femur bone mass more effectively than spinal bone mass. [1] Balicatib is a cathepsin K inhibitor developed for the treatment of osteoporosis. Unlike most bone resorption inhibitors (e.g., bisphosphonates), Balicatib demonstrated a unique ability to stimulate periosteal bone formation in this long-term primate study, suggesting it may have anabolic properties in addition to its anti-resorptive effects. [1] The study suggests that the stimulatory effect on periosteal bone formation, combined with weak inhibition of endocortical bone formation in the femur, may explain why Balicatib increased femur bone mass more effectively than spinal bone mass. [1] |
Solubility Data
| Solubility (In Vitro) |
DMSO: 75~82 mg/mL (182.2~199.3 mM) Ethanol: ~3 mg/mL (~7.3 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 3.75 mg/mL (9.11 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 37.5 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 3.75 mg/mL (9.11 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 37.5 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4299 mL | 12.1495 mL | 24.2990 mL | |
| 5 mM | 0.4860 mL | 2.4299 mL | 4.8598 mL | |
| 10 mM | 0.2430 mL | 1.2149 mL | 2.4299 mL |