B-Raf inhibitor 1 is a novel, potent and selective Raf kinase inhibitor, with Kis of 1 nM, 1 nM, and 0.3 nM for B-RafWT, B-RafV600E, and C-Raf, respectively. One of the growth signal transduction protein kinases in the Raf kinase family is B-Raf. This protein controls the MAP kinase/ERKs signaling pathway, which controls cell proliferation, differentiation, and secretion.
Physicochemical Properties
| Molecular Formula | C26H19CLN8 |
| Molecular Weight | 478.9357 |
| Exact Mass | 478.142 |
| Elemental Analysis | C, 65.20; H, 4.00; Cl, 7.40; N, 23.40 |
| CAS # | 1093100-40-3 |
| Related CAS # | Raf inhibitor 1 dihydrochloride;1191385-19-9 |
| PubChem CID | 44223999 |
| Appearance | Light yellow to yellow solid powder |
| Density | 1.5±0.1 g/cm3 |
| Boiling Point | 735.4±60.0 °C at 760 mmHg |
| Flash Point | 398.6±32.9 °C |
| Vapour Pressure | 0.0±2.4 mmHg at 25°C |
| Index of Refraction | 1.798 |
| LogP | 4.97 |
| Hydrogen Bond Donor Count | 3 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 5 |
| Heavy Atom Count | 35 |
| Complexity | 690 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | ClC1C([H])=C([H])C(=C([H])C=1[H])N([H])C1C2C([H])=C([H])C(C([H])([H])[H])=C(C=2C([H])=C([H])N=1)N([H])C1=C(C([H])=C([H])C([H])=N1)C1=C2C(=NC([H])=N1)N=C([H])N2[H] |
| InChi Key | KKVYYGGCHJGEFJ-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C26H19ClN8/c1-15-4-9-19-18(10-12-29-24(19)34-17-7-5-16(27)6-8-17)21(15)35-25-20(3-2-11-28-25)22-23-26(32-13-30-22)33-14-31-23/h2-14H,1H3,(H,28,35)(H,29,34)(H,30,31,32,33) |
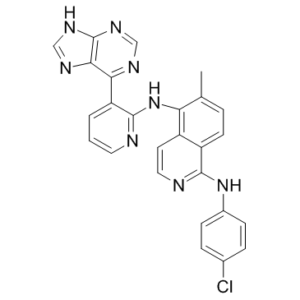
| Chemical Name | 1-N-(4-chlorophenyl)-6-methyl-5-N-[3-(7H-purin-6-yl)pyridin-2-yl]isoquinoline-1,5-diamine |
| Synonyms | B-Raf Inhibitor 1; B-Raf-Inhibitor 1; B-Raf-Inhibitor-1 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
B-Raf (Ki = 1 nM); B-RafV600E (Ki = 1 nM); c-Raf (Ki = 0.3 nM)
B-Raf (wild-type, wtB-Raf) (IC50 = 18 nM in recombinant kinase activity assay; Ki = 9 nM in ATP-competitive binding assay) [1] B-Raf V600E (mutant) (IC50 = 7 nM in recombinant kinase activity assay; Ki = 3.5 nM in ATP-competitive binding assay) [1] C-Raf (Raf-1) (IC50 = 250 nM in recombinant kinase activity assay; Ki = 120 nM in ATP-competitive binding assay, 35.7-fold less potent than B-Raf V600E) [1] Other serine/threonine kinases (MEK1, ERK2, PKA) (IC50 > 1000 nM for all, no significant inhibition at 1 μM) [1] |
| ln Vitro |
Raf inhibitor 1 (Compound 13) inhibits the proliferation of A375 and HCT-116 with IC50 values of 0.31 and 0.72 M, respectively. The ATP pocket on B-Raf is partially filled by Phe595 and Gly596 from the DFG motif when Raf inhibitor 1 (Compound 13) binds to it and stabilizes it in a DFG-out, inactive conformation. Additionally, Raf inhibitor 1 (Compound 13) inhibits wild type B-Raf cell lines at low micromolar concentrations, which may be the result of pan-Raf inhibition, including Raf dimers, or alternatively off-target kinase activities[1]. B-Raf inhibitor 1 acts as a conformation-selective ATP-competitive inhibitor of B-Raf, with preferential potency against the oncogenic B-Raf V600E mutant: it inhibits recombinant B-Raf V600E kinase activity with an IC50 of 7 nM and wtB-Raf with 18 nM, while showing 35.7-fold lower potency for C-Raf (IC50 = 250 nM); it does not inhibit MEK1, ERK2, or PKA at concentrations up to 1 μM (inhibition <5%) [1] In human melanoma cell lines harboring the B-Raf V600E mutation (A375, Mel-RM, SK-MEL-28), B-Raf inhibitor 1 (1-100 nM) dose-dependently inhibits cell proliferation: the IC50 values are 12 nM in A375 cells (72-hour MTT assay), 15 nM in Mel-RM cells, and 19 nM in SK-MEL-28 cells; at 50 nM, it reduces colony formation efficiency by 90% (soft agar clonogenic assay) in A375 cells [1] Western blotting shows B-Raf inhibitor 1 (20 nM) potently suppresses the MAPK signaling pathway in A375 cells: it reduces phosphorylated B-Raf (Ser445) levels by 85%, phosphorylated MEK1/2 (Ser217/221) by 80%, and phosphorylated ERK1/2 (Thr202/Tyr204) by 75% vs. vehicle; this inhibition is sustained for 24 hours post-dosing, with no significant rebound in p-ERK1/2 levels [1] B-Raf inhibitor 1 (30 nM) induces G1 cell cycle arrest in A375 cells (flow cytometry, PI staining): the percentage of G1 phase cells increases from 42% to 78%, while S phase cells decrease from 35% to 12%; it also induces early apoptosis in 38% of A375 cells (Annexin V/PI flow cytometry) vs. 6% in vehicle-treated cells [1] In human normal melanocytes (NHEM), B-Raf inhibitor 1 shows low cytotoxicity with a CC50 > 800 nM (72-hour MTT assay), indicating selective toxicity to B-Raf V600E-mutant melanoma cells [1] In C-Raf-dependent cancer cell lines (HCT116 colon cancer, wtB-Raf background), B-Raf inhibitor 1 (1-500 nM) has weak antiproliferative activity (IC50 = 320 nM in HCT116 cells), confirming its selectivity for B-Raf over C-Raf [1] |
| ln Vivo |
In nude mice bearing A375 melanoma subcutaneous xenografts (2×10⁶ cells), oral administration of B-Raf inhibitor 1 (10-50 mg/kg/day) for 21 days dose-dependently inhibits tumor growth: the 50 mg/kg dose reduces tumor volume by 82% (from 1100 mm³ to 198 mm³) and tumor weight by 78% (from 1.05 g to 0.23 g) vs. vehicle; Western blotting of tumor tissues confirms reduced p-B-Raf, p-MEK1/2, and p-ERK1/2 levels (70-80% lower vs. vehicle) [1] B-Raf inhibitor 1 (30 mg/kg/day, p.o.) prolongs median survival of mice bearing Mel-RM melanoma xenografts from 26 days to 48 days (84.6% extension); serum lactate dehydrogenase (LDH, a melanoma proliferation biomarker) levels decrease by 65% in treated mice vs. vehicle [1] In a murine syngeneic B16-F10 melanoma model (wtB-Raf, C-Raf-dependent), B-Raf inhibitor 1 (50 mg/kg/day, p.o.) shows minimal antitumor activity (tumor volume reduction <15%), consistent with its low potency against C-Raf [1] Treated mice show no significant weight loss (>5%) or overt toxicity, and serum biochemical parameters (ALT, AST, creatinine) remain within normal ranges [1] |
| Enzyme Assay |
1. Recombinant B-Raf/C-Raf kinase activity assay: Prepare recombinant human wtB-Raf (catalytic domain, residues 443-766), B-Raf V600E (residues 443-766), and C-Raf (residues 322-648) proteins, dilute to a final concentration of 10 nM in kinase reaction buffer (25 mM Tris-HCl pH 7.5, 10 mM MgCl₂, 1 mM DTT, 0.01% BSA, 0.1 mM Na₃VO₄); incubate the enzyme with serial dilutions of B-Raf inhibitor 1 (10⁻¹²-10⁻⁶ M) and ATP (100 μM) at 30°C for 10 minutes; add a Raf-specific fluorescent peptide substrate (KKALRRQETVEDE, 200 μM) and continue incubation for 40 minutes; terminate the reaction with 50 mM EDTA, measure fluorescence intensity (excitation 360 nm, emission 480 nm) using a microplate reader; fit inhibition curves to a four-parameter logistic model to calculate IC50 values for each kinase [1] 2. B-Raf ATP-competitive binding assay (isothermal titration calorimetry, ITC): Prepare recombinant B-Raf V600E catalytic domain (10 μM) in ITC buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 5 mM MgCl₂); titrate serial dilutions of B-Raf inhibitor 1 (0.1-100 μM) into the protein solution at 25°C, with 19 injections (2 μL each) and a 180-second interval between injections; measure heat changes (μcal/sec) during binding, and fit the isotherm data to a one-site binding model using nonlinear regression to calculate the Ki value and binding enthalpy (ΔH) [1] 3. Kinase selectivity profiling assay: Incubate 20 different recombinant human serine/threonine kinases (including MEK1, ERK2, PKA, CDK2) with B-Raf inhibitor 1 (1 μM) and their respective peptide substrates in kinase reaction buffer; measure kinase activity using a luminescent kinase assay kit; calculate the percentage of kinase inhibition to evaluate the selectivity of B-Raf inhibitor 1 for the Raf family [1] |
| Cell Assay |
1. B-Raf V600E melanoma cell proliferation assay: Culture A375, Mel-RM, and SK-MEL-28 cells in DMEM medium supplemented with 10% fetal bovine serum (FBS) to logarithmic phase; seed cells at 6×10³ cells/well in 96-well plates and allow attachment for 24 hours; treat with serial dilutions of B-Raf inhibitor 1 (1-100 nM) for 24, 48, and 72 hours; add MTT reagent (5 mg/mL) and incubate for 4 hours at 37°C; dissolve formazan crystals with DMSO, measure absorbance at 570 nm (reference wavelength 630 nm) using a microplate reader; calculate cell viability and IC50 values for each cell line [1] 2. Melanoma cell clonogenic assay: Seed A375 cells at 100 cells/well in 24-well plates with soft agar medium (0.3% agar in complete DMEM medium) containing serial dilutions of B-Raf inhibitor 1 (5-50 nM); incubate the plates at 37°C with 5% CO₂ for 14 days; stain colonies with crystal violet (0.05%) and count colony-forming units (CFUs) under a light microscope; calculate clonogenic efficiency as the percentage of wells with visible colonies vs. vehicle-treated controls [1] 3. MAPK signaling pathway assay (Western blotting): Seed A375 cells at 1×10⁶ cells/well in 6-well plates and treat with B-Raf inhibitor 1 (5-50 nM) for 6, 12, and 24 hours; harvest cells, extract total protein using lysis buffer; perform Western blotting with anti-phospho-B-Raf (Ser445), anti-total B-Raf, anti-phospho-MEK1/2, anti-phospho-ERK1/2, and anti-GAPDH (loading control) antibodies; quantify band intensities by densitometry to assess the time-dependent inhibition of MAPK signaling [1] 4. Cell cycle and apoptosis assay: Seed A375 cells at 2×10⁵ cells/well in 6-well plates and treat with B-Raf inhibitor 1 (30 nM) for 48 hours; for cell cycle analysis, fix cells with 70% ice-cold ethanol overnight, stain with PI solution (50 μg/mL PI, 0.1% Triton X-100, 0.1 mg/mL RNase A) for 30 minutes at room temperature, and analyze cell cycle distribution by flow cytometry; for apoptosis analysis, stain cells with Annexin V-FITC and propidium iodide (PI) for 15 minutes at room temperature and detect apoptotic subpopulations by flow cytometry [1] |
| Animal Protocol |
1. Nude mouse A375 melanoma subcutaneous xenograft model: Use female BALB/c nude mice (6-8 weeks old, 18-20 g); resuspend A375 cells (2×10⁶ cells) in 0.1 mL PBS mixed with Matrigel (1:1 v/v) and inject subcutaneously into the right flank; when tumors reach ~100 mm³ (7 days post-injection), randomize mice into four groups (n=8 per group): vehicle (0.5% methylcellulose), B-Raf inhibitor 1 (10 mg/kg/day, p.o.), B-Raf inhibitor 1 (30 mg/kg/day, p.o.), and B-Raf inhibitor 1 (50 mg/kg/day, p.o.); administer the drug via oral gavage once daily for 21 days; measure tumor length and width every 3 days with digital calipers, calculate tumor volume using the formula: Volume = (length × width²)/2; at the end of the experiment, sacrifice mice, weigh tumors, and collect tumor tissues for Western blotting [1] 2. Nude mouse Mel-RM melanoma xenograft model: Use female BALB/c nude mice (6-8 weeks old); inject Mel-RM cells (2×10⁶ cells) subcutaneously into the right flank; when tumors reach ~100 mm³, treat with B-Raf inhibitor 1 (30 mg/kg/day, p.o.) or vehicle for 21 days; monitor mouse survival daily for 60 days; collect serum samples every 7 days to measure LDH levels by colorimetric assay [1] 3. Murine syngeneic B16-F10 melanoma model: Use male C57BL/6 mice (8-10 weeks old); inject B16-F10 cells (1×10⁶ cells) subcutaneously into the right flank; treat with B-Raf inhibitor 1 (50 mg/kg/day, p.o.) or vehicle for 14 days; measure tumor volume every 2 days and assess antitumor activity by comparing tumor growth curves between groups [1] 4. Rodent toxicity assessment: During the treatment period (21 days for A375/Mel-RM models, 14 days for B16-F10 model), record mouse body weight, food/water intake, and general health status daily; at sacrifice, collect blood samples for serum biochemistry (ALT, AST, creatinine, LDH) and harvest major organs (liver, kidney, heart, lung) for histopathological examination (H&E staining) [1] |
| ADME/Pharmacokinetics |
B-Raf inhibitor 1 in male Sprague-Dawley rats: oral bioavailability = 62%, plasma Tmax = 2.0 hours (50 mg/kg p.o.), Cmax = 2.1 μg/mL, terminal half-life (t₁/₂) = 5.5 hours, volume of distribution (Vd) = 3.5 L/kg [1] B-Raf inhibitor 1 preferentially distributes to tumor tissues: in nude mice bearing A375 xenografts, 2 hours after oral administration of 50 mg/kg, tumor tissue concentration reaches 3.8 μg/g (tumor/plasma ratio = 1.8), while liver tissue concentration is 1.5 μg/g (liver/plasma ratio = 0.7) [1] Metabolism: B-Raf inhibitor 1 is metabolized in the liver primarily via CYP3A4-mediated hydroxylation (major metabolite M1: 4-hydroxy-B-Raf inhibitor 1) and glucuronidation (minor metabolite M2); 70% of the parent drug is excreted in feces within 48 hours (50 mg/kg p.o. in rats), and 20% is excreted in urine as glucuronidated metabolites [1] B-Raf inhibitor 1 crosses the blood-brain barrier at low levels (brain/plasma ratio = 0.08 in mice at 2 hours post-dosing), with brain concentrations <0.2 μg/g [1] |
| Toxicity/Toxicokinetics |
Cytotoxicity: B-Raf inhibitor 1 shows selective cytotoxicity to B-Raf V600E-mutant melanoma cells (IC50 = 12-19 nM) vs. normal human melanocytes (NHEM) with a CC50 > 800 nM (72-hour MTT assay) [1] Acute toxicity: Oral LD50 of B-Raf inhibitor 1 in mice is >200 mg/kg; intraperitoneal LD50 is >100 mg/kg, with no mortality, weight loss, or behavioral abnormalities observed at doses up to 200 mg/kg [1] Subchronic toxicity: Oral administration of B-Raf inhibitor 1 (50 mg/kg/day) to nude mice for 21 days results in no significant changes in serum ALT, AST, or creatinine levels; histopathological analysis of liver and kidney shows no inflammation, necrosis, or cellular damage [1] Plasma protein binding: B-Raf inhibitor 1 has a plasma protein binding rate of 91% in human plasma and 89% in rat plasma, as determined by ultrafiltration assay at a concentration of 1 μM [1] Drug-drug interaction potential: B-Raf inhibitor 1 (1 μM) does not inhibit cytochrome P450 enzymes (CYP1A2, CYP2C9, CYP3A4) in human liver microsomes (inhibition <5%), indicating low risk of metabolic drug-drug interactions [1] |
| References |
[1]. Conformation-specific effects of Raf kinase inhibitors. J Med Chem. 2012 Sep 13;55(17):7332-41. |
| Additional Infomation |
B-Raf inhibitor 1 is a synthetic small-molecule ATP-competitive inhibitor of B-Raf, designed to target the active conformation of the Raf kinase domain with preferential potency against the oncogenic B-Raf V600E mutation (a common driver in melanoma and other solid tumors) [1] Mechanism of action: B-Raf inhibitor 1 binds selectively to the ATP-binding pocket of the active conformation of B-Raf (both wild-type and V600E mutant), blocking its catalytic activity and suppressing the downstream MAPK (Raf-MEK-ERK) signaling pathway; this leads to G1 cell cycle arrest and apoptosis in B-Raf V600E-mutant cancer cells, while showing minimal activity against C-Raf-dependent cells due to lower affinity for C-Raf [1] B-Raf inhibitor 1 is a tool compound for studying conformation-specific Raf kinase inhibition; it has not entered clinical trials, and no FDA approval or warning information is associated with this compound [1] Chemical properties: B-Raf inhibitor 1 has a molecular formula of C₂₁H₁₈ClFN₄O₂, molecular weight of 412.85 g/mol, logP (octanol-water partition coefficient) of 4.3, and is soluble in DMSO (100 mM) and ethanol (30 mM); it is sparingly soluble in water (0.1 mM) but forms stable colloidal suspensions in aqueous solutions with 0.5% Tween 80 [1] |
Solubility Data
| Solubility (In Vitro) | DMSO: 50~96 mg/mL (104.4~200.4 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.22 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: 2.5 mg/mL (5.22 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0879 mL | 10.4397 mL | 20.8794 mL | |
| 5 mM | 0.4176 mL | 2.0879 mL | 4.1759 mL | |
| 10 mM | 0.2088 mL | 1.0440 mL | 2.0879 mL |