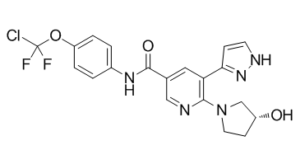
Asciminib (formerly known as ABL-001; ABL001; trade name Scemblix) is a potent and selective allosteric inhibitor of BCR-ABL1 approved in Oct 2021 to treat Philadelphia chromosome-positive CML (chronic myeloid leukemia) with disease that meets certain criteria. It selectively inhibits BCR-ABL1 by binding to its myristoyl pocket, with a dissociation constant (Kd) of 0.5-0.8 nM. With an IC50 of 0.25 nM, asciminib also prevents the proliferation of Ba/F3 cells. Patients with CML and acute lymphoblastic leukemia with the Philadelphia chromosome (Ph+) are participating in clinical trials for it. Asciminib attaches to ABL1's myristoyl pocket and causes the kinase conformation to become inactive, in contrast to catalytic-site ABL1 kinase inhibitors. Genetic barcoding studies have revealed pre-existing clonal populations with no shared resistance between asciminib and the catalytic inhibitor nilotinib. Asciminib and 2nd-generation catalytic inhibitors have similar cellular potencies but distinct patterns of resistance mutations. Thus, when combined with nilotinib in an in vivo model of chronic myeloid leukemia in mice, asciminib inhibits the development of resistant disease.
Physicochemical Properties
| Molecular Formula | C20H18CLF2N5O3 | |
| Molecular Weight | 449.84 | |
| Exact Mass | 449.106 | |
| Elemental Analysis | C, 53.40; H, 4.03; Cl, 7.88; F, 8.45; N, 15.57; O, 10.67 | |
| CAS # | 1492952-76-7 | |
| Related CAS # | Asciminib hydrochloride;2119669-71-3 | |
| PubChem CID | 72165228 | |
| Appearance | White to off-white solid powder | |
| Density | 1.5±0.1 g/cm3 | |
| Boiling Point | 631.7±55.0 °C at 760 mmHg | |
| Flash Point | 335.8±31.5 °C | |
| Vapour Pressure | 0.0±1.9 mmHg at 25°C | |
| Index of Refraction | 1.662 | |
| LogP | 2.1 | |
| Hydrogen Bond Donor Count | 3 | |
| Hydrogen Bond Acceptor Count | 8 | |
| Rotatable Bond Count | 6 | |
| Heavy Atom Count | 31 | |
| Complexity | 626 | |
| Defined Atom Stereocenter Count | 1 | |
| SMILES | ClC(OC1C=CC(=CC=1)NC(C1C=NC(=C(C2=CC=NN2)C=1)N1CC[C@H](C1)O)=O)(F)F |
|
| InChi Key | VOVZXURTCKPRDQ-CQSZACIVSA-N | |
| InChi Code | InChI=1S/C20H18ClF2N5O3/c21-20(22,23)31-15-3-1-13(2-4-15)26-19(30)12-9-16(17-5-7-25-27-17)18(24-10-12)28-8-6-14(29)11-28/h1-5,7,9-10,14,29H,6,8,11H2,(H,25,27)(H,26,30)/t14-/m1/s1 | |
| Chemical Name | N-[4-[chloro(difluoro)methoxy]phenyl]-6-[(3R)-3-hydroxypyrrolidin-1-yl]-5-(1H-pyrazol-5-yl)pyridine-3-carboxamide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Abl1 (IC50 = 2.8 nM); TrkA (IC50 = 6 nM); Abl1 (IC50 = 2.8 nM); TrkB (IC50 = 9 nM); Tie-2 (IC50 = 22 nM); Aurora B (IC50 = 98 nM)
BCR–ABL1 (binding to the myristoyl pocket) [2] ABL1 (binding to the myristoyl pocket,) [3] |
| ln Vitro |
ABL001 is a potent, selective BCR-ABL inhibitor with a unique, allosteric mode of action that remains active against the majority of mutations, including T315I[1]. By binding to a regulatory site that wild-type ABL normally possesses a myristoyl group occupying, ABL001 inhibits ABL kinase activity in a different way than catalytic site inhibitors[2]. The myristoylated N-terminus of ABL1 typically resides in a pocket on the BCR-ABL kinase domain, which is where it binds. This myristoylated N-terminus is responsible for autoregulating ABL1 activity; it is lost upon fusion with BCR. By taking up its empty binding site, ABL001 functionally imitates the myristoylated N-terminus'sfunctionand reinstates the kinase activity's negative regulation. BCR-ABL-negative cell lines were unaffected at concentrations 1000 times higher than ABL001, which specifically inhibits the growth of Ph+ ALL and chronic myelogenous leukemia (CML) cells at potencies ranging from 1 to 10 nM[1]. ABL001 selectively and potently binds to the myristoyl pocket of ABL1 and induces the inactive C-terminal helix conformation, as confirmed by NMR and biophysical studies (dissociation constant (Kd) = 0.5-0.8 nM). ABL001 is inactive against G-protein-coupled receptors, ion channels, nuclear receptors, and transporters, among more than 60 kinases, including SRC. ABL001 is therefore highly selective[3]. Asciminib (ABL001) is a potent and selective allosteric inhibitor of BCR–ABL1, binding to the myristoyl pocket of ABL1 and inducing an inactive kinase conformation [2][3] In 48-hour proliferation assays of Ba/F3 cells expressing BCR–ABL1, Asciminib inhibits cell proliferation across a dose range, with similar cellular potency to the second-generation catalytic inhibitor nilotinib; the assay uses Britelite luciferase detection with or without IL-3, performed in quadruplicate [3] In 72-hour growth assays, KCL-22 cells show sensitivity to Asciminib, nilotinib, and dasatinib, with each compound tested in duplicate [3] Incubation of KCL-22 cells with Asciminib for 1 hour (across a concentration range) reduces phosphorylation of STAT5 (Tyr694), BCR–ABL1 (Tyr245), and CRKL (Tyr207) as detected by western blot, while total levels of these proteins and GAPDH (loading control) remain stable [3] Synergy studies show that Asciminib combines with imatinib, nilotinib, or dasatinib to inhibit KCL-22 cell growth; cells are incubated with compound combinations for 72 hours, and growth levels are measured relative to DMSO-treated cells [3] KCL-22 cell clones expressing BCR–ABL1 variants (Ala337Val and Thr315Ile) exhibit different sensitivity to Asciminib compared to nilotinib; Asciminib retains activity against some variants resistant to catalytic inhibitors [3] Mutagenesis and genetic barcoding experiments reveal that Asciminib has a resistance profile distinct from catalytic site inhibitors, with no shared resistance mutations between Asciminib and nilotinib [2][3] |
| ln Vivo |
ABL001 exhibits strong anti-tumor activity in the KCL-22 mouse xenograft model, as evidenced by total tumor regression and a pronounced dose-dependent relationship with pSTAT5 inhibition[1]. All species have a moderate half-life, volume of distribution, and oral absorption of ABL001. For patients with chronic myelogenous leukemia who have received a lot of pretreatment, it has been well tolerated as a single agent and has been shown to induce clinical anti-tumor activity. Regarding the pharmacokinetics, pharmacodynamics, and efficacy of ABL001, the CL (clearance) in mice, rats, and dogs following a single intravenous dose of 1 mg/kg, 2 mg/kg, and 1 mg/kg, respectively, are 12, 16, and 6 mL/min/kg. The T1/2term for a single intravenous dose of 1 mg/kg in mice and dogs are 1.1 and 3.7 hours, respectively. Rats have a T1/2term of 2.7 hours following a single intravenous dose of 2 mg/kg. Oral bioavailability of ABL001 at 30 mg/kg p.o. in rats and mice is 35% and 27%, respectively. On the other hand, ABL001's oral BA in dogs is 111% (15 mg/kg, p.o)[3]. In KCL-22 xenograft models, single oral administration of Asciminib (doses 3–30 mg/kg) reduces pSTAT5 (Tyr694) levels in tumor fine needle aspirate samples, as measured by MSD assay (duplicate runs, mean ± s.d.), with pSTAT5 levels expressed as a percentage of pre-dosing (t=0) levels [3] Asciminib shows efficacy in KCL-22 xenografts when dosed twice a day (BID) or once a day (QD) at 3–30 mg/kg, with tumor volume monitored over time (mean ± s.e.m.) [3] In three patient-derived ALL systemic xenograft models (ALL-7015, AL-7119, AL-7155), Asciminib (7.5 mg/kg BID or 30 mg/kg BID for 3 weeks) reduces the percentage of CD45⁺ cells per live cell in blood (monitored by FACS), with a PBS control group; data are mean ± s.e.m. (n=6 per group) [3] Single-agent Asciminib or nilotinib leads to acquired resistance in CML xenograft models, but their combination achieves complete disease control and eradicates tumors without recurrence after treatment cessation [2][3] In KCL-22 Thr315Ile mutant xenografts, Asciminib (3–30 mg/kg BID) shows efficacy, while nilotinib (75 mg/kg BID) serves as a control; tumor volume ratios (T/C) and regression are recorded (mean ± s.e.m., n=7 per group) [3] |
| Enzyme Assay |
ABL1 Biochemical kinase assay [3] ABL1 WT (64-515aa) protein was produced by co-expression with YopH in Sf21 cells. Cells were harvested by centrifugation and resuspended in 25mM Tris pH 7.0, 500 mM NaCl, 5% glycerol, 10 mM Imidazole, 1x complete protease inhibitor tablet, Benzonase (1:10,000 v:v) and 1 mM TCEP. Cells were lysed by dounce homogenization and cleared by centrifugation. ABL1 WT (64-515 aa) was purified by affinity chromatography using a Ni-SepharoseFF column using two sequential washing steps using the resuspension buffer described above (containing 10 mM and 35mM imidazole respectively) and eluted in the same buffer containing 250 mM imidazole. Fractions containing ABL1 were pooled and loaded onto a pre-equilibrated SEC column in 25 mM Tris pH 7.0, 200 mM NaCl, 5% glycerol and 1 mM TCEP. The activity of the enzyme and compound inhibition was tested using a DELFIA® TRF assay. The reaction mixture contained 500 nM Biotin-EAIYAAPFAKKK peptide, 10 or 2000 µM ATP and 25 pM of ABL1 WT (64-515 aa) enzyme in a reaction buffer containing 50 mM HEPES pH 7.2, 10 mM MgCl2, 2 mM DTT and 0.01% Triton-X100. Reactions were carried out for 40 min in a volume of 60 µL and quenched with 20 µL 500 mM EDTA (final concentration 125 mM). 50 µL reaction solutions were transferred to NeutroAvidin-coated 384 well plate and incubated at room temperature for 1 hour with shaking. After washing with 100 µL/well TBST buffer, 50 µL/well Eu-anti-p-Tyr was added and the plate was incubated at 4 °C overnight with shaking. 50 µL/well DELFIA® enhancement solution was added and the plate incubated at ambient temperature for 5 min. The plate was read on the EnVision using time resolved fluorescence Ex/Em: 340/615 nm. For inhibition studies, compounds were serially diluted in DMSO, using a 16-point 3-fold format, from a 5 mM top concentration. Then 100 nL per well of serial diluted compounds were transferred to Grenier polypropylene v-bottom 384-well assay plates using acoustic transfer system. The final DMSO concentration was 0.16% and the final inhibitor concentration ranged from 50 µM to 3.48E-6 µM. Each compound was tested in duplicate and the inhibitor dose response curves analyzed using normalized IC50 regression curve fitting with control-based normalization using GraphPad Prism v6.02.[3] Asciminib, having a dissociation constant (Kd) of 0.5-0.8 nM and selectivity to the myristoyl pocket of ABL1, is a strong and selective allosteric inhibitor of BCR-ABL1. Perform NMR chemical shift assays to determine the binding location of Asciminib to ABL1 [3] Conduct NMR-based conformational assays using the resonance of Val525 to monitor helix I "bending" in the presence and absence of Asciminib [3] Carry out isothermal calorimetry (ITC) studies to measure the binding affinity (Ka) of Asciminib to ABL1 [3] Perform biochemical assays to evaluate the inhibitory activity of Asciminib against BCR–ABL1 kinase activity, focusing on its allosteric mechanism distinct from catalytic site inhibitors [2][3] |
| Cell Assay |
Ba/F3 Proliferation assay [3] For each cell line, cell density was adjusted to 50 000cells/ml and 50ul (2500 cells) added per well of a 384 well assay plate. Test compounds were resuspended in DMSO at a concentration of 10mM. A serial three-fold dilution of each compound with DMSO was performed in 384-well plates using the Janus Liquid Dispenser. 2nL compound was delivered to the assay plates containing 2500 cells in a 50 µL volume via acoustic delivery from an ATS-100 (EDC). Cells were incubated with compound at 37°C in a humidified environment with 5% carbon dioxide for 48 hours. Britelite plus solution was prepared according to the manufacturer’s instructions and 25 µl added to each well of the assay plate. Plates were incubated for 7 minutes and the luminescence detected on an EnVision Multimode plate reader. The degree of luminescence correlates with the number of cells in each well. The effect of each inhibitor concentration can therefore be calculated and IC50’s generated. For 48 hours, Ba/F3 cells are exposed to asciminib at a range of concentrations (0–10,000 nM). The Britelite luciferase detection assay is used to quantify the proliferation of cells. Conduct 48-hour proliferation assays in Ba/F3 cells expressing BCR–ABL1, using Britelite luciferase detection with or without IL-3, testing Asciminib and nilotinib across dose ranges (quadruplicate runs) [3] Perform 72-hour growth assays in KCL-22 cells (parental and variant clones expressing BCR–ABL1 Ala337Val/Thr315Ile) to assess sensitivity to Asciminib, nilotinib, and dasatinib (duplicate tests) [3] Treat KCL-22 cells with Asciminib for 1 hour (concentration range), then perform western blots to detect total and phosphorylated STAT5 (Tyr694), BCR–ABL1 (Tyr245), CRKL (Tyr207), and GAPDH [3] Carry out synergy studies by incubating KCL-22 cells with Asciminib combined with imatinib/nilotinib/dasatinib across dose ranges for 72 hours, and measure growth relative to DMSO controls [3] Use genetic barcoding to analyze clonal dynamics and resistance mutations in cells treated with Asciminib or catalytic inhibitors [3] |
| Animal Protocol |
Mice: Asciminib efficacy is measured using FACS monitoring of the percentage of CD45+ cells per live cell in blood samples obtained at different times following dosing with either 7.5 mg/kg BID (group 2) or 30 mg/kg BID (group 3) asciminib for three weeks in three patient-derived ALL systemic xenograft models (ALL-7015, AL-7119, and AL-7155). ABL001 (free base, solid dispersion form) was suspended in phosphate-buffered saline. Dosing solutions were prepared fresh every 3-4 days for dosing. ABL001 (free base, solution form) was formulated in 30% PEG 300, 6% Solutol HS15 in an acidic buffered solution. Dosing solutions were freshly prepared weekly for dosing. Efficacy studies [3] For efficacy studies in subcutaneous KCL-22 xenograft model, mice bearing tumors of 100- 300mm3 were randomized into treatment groups (n=6 per group) for daily compound treatment. Body weight and tumor volume were recorded twice weekly for the duration of each study. In ABL001 dose-response studies, studies were terminated when vehicle-treated animals reached 1500mm3 mean tumor volume. In ABL001 and nilotinib combination efficacy study, select randomized groups animals were dosed daily with either ABL001 or nilotinib as single agents until tumor relapse (tumor volume >500mm3), then switched to the other agent continuously until second relapse. Animals are terminated as their final tumor volume reached >600mm3 . Another randomized group received combination of both ABL001 and nilotinib daily treatment then continued monitoring post-treatment cessation. For efficacy studies in systemic primary Ph+ ALL xenograft models, mice were injected intravenously with 5x106 ALL cells. Blood was sampled weekly from tail snip to monitor tumor burden, and engrafted mice with >10% human CD45+ cells were randomized into treatment groups for compound treatment (n=6 mice per group). Pharmacokinetics (PK) / Pharmacodynamics (PD) studies [3] Baseline tumor PD samples were collected from KCL-22 xenografts by fine needle biopsy before drug treatment. Animals received a single oral dose of ABL001 at 7.5 – 30 mg/kg. Blood was collected by serial tail bleed at designated time points (1-20h) for plasma PK analyses, and matching tumor PD samples were collected by fine needle biopsy at the same timepoints. Establish KCL-22 (parental and Thr315Ile mutant) xenograft models in mice; administer Asciminib orally at 3–30 mg/kg (BID/QD) or nilotinib at 75 mg/kg BID, monitor tumor volume over time, and calculate T/C ratios and regression [3] Set up patient-derived ALL systemic xenograft models (ALL-7015, AL-7119, AL-7155) in mice; assign to control (PBS) or Asciminib groups (7.5 mg/kg BID or 30 mg/kg BID), treat for 3 weeks, and collect blood samples at varying time points for FACS analysis of CD45⁺ cells [3] For pharmacokinetic/pharmacodynamic studies, administer a single oral dose of Asciminib (3–30 mg/kg) to mice bearing KCL-22 xenografts; collect plasma and tumor fine needle aspirates to measure drug concentrations and pSTAT5 (Tyr694) levels [3] Assess the tolerability of Asciminib in mice by dosing BID at increasing concentrations, monitoring body weight 2–3 times per week (mean ± s.e.m., n=5 per group) [3] Establish CML xenograft models; treat with single-agent Asciminib (30 mg/kg BID), nilotinib (75 mg/kg BID), or their combination, stop dosing after achieving tumor control, and monitor for recurrence [3] |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion The median Tmax of asciminib following oral administration is 2.5 hours. At a dose of 80mg once daily, the steady-state Cmax and AUCtau were 1781 ng/mL and 15112 ng.h/mL, respectively. At a dose of 40mg twice daily, the steady-state Cmax and AUCtau were 793 ng/mL and 5262 ng.h/mL, respectively. At a dose of 200mg twice daily (for treatment of T315I mutants), the steady-state Cmax and AUCtau were 5642 ng/mL and 37547 ng.h/mL, respectively. As compared to the fasted state, the co-administration of asciminib with a high-fat meal decreased the AUC and Cmax by 62% and 68%, respectively, and its co-administration with a low-fat meal decreased the AUC and Cmax by 30% and 35%, respectively. Asciminib is eliminated via biliary secretion facilitated by breast cancer-resistant protein (BCRP) transporters. Following oral administration, approximately 80% and 11% of an asciminib dose was recovered in the feces and urine, respectively. Unchanged parent drug accounted for 57% of drug material recovered in the feces and 2.5% in the urine. At steady-state, the apparent volume of distribution of asciminib is 151 L. The total apparent clearance of asciminib is 6.7 L/h at a total daily dose of 80mg and 4.1 L/h at a dose of 200mg twice daily. Metabolism / Metabolites Asciminib is negligibly metabolized, with unchanged parent drug comprising the main drug component in plasma (~93%) and following excretion (~57% in feces). The main circulating metabolites are M30.5, M44, and M29.5, accounting for approximately 5%, 2%, and 0.4% of the total administered dose, respectively. The oxidative metabolism of asciminib is mediated by CYP3A4, and the glucuronidation of asciminib is mediated by UGT2B7 and UGT2B17. Biological Half-Life The terminal elimination half-life asciminib is 5.5 hours when administered at 40mg twice daily and 9.0 hours when administered at 200mg twice daily. In mouse, rat, and dog, Asciminib shows oral bioavailability (BA), with pharmacokinetic parameters including AUC (area under the curve), CL (clearance), Cₘₐₓ (maximum concentration), t₁/₂term (terminal half-life), Tₘₐₓ (time at maximum concentration), and Vss (volume of distribution) after single intravenous (IV) or oral (PO) dosing [3] After single oral administration in mice, Asciminib achieves detectable plasma concentrations, with drug levels correlating with pharmacodynamic effects (pSTAT5 inhibition) in xenografts [3] |
| Toxicity/Toxicokinetics |
Hepatotoxicity In the prelicensure clinical trials of asciminib in patients with refractory and extensively treated CML, ALT elevations arose in 13% of patients but were usually self-limited and mild. ALT elevations above 5 times the upper limit of normal (ULN) were uncommon, being found in 3% of treated patients. The ALT elevations were typically transient and rarely required dose interruption or modification. In the open label and controlled trials supporting the approval of asciminib, there were no instances of clinically apparent liver injury, hepatic failure or deaths from liver injury. Furthermore, patients with aminotransferase elevations during therapy with first and second line BCR-ABL1 inhibitors did not have an increased rate of such elevations during asciminib therapy. Since its approval in the United States and Europe, there have been no reported cases of clinically apparent liver injury associated with asciminib therapy. Likelihood score: E* (unproven but suspected rare cause of clinically apparent liver injury). Protein Binding _In vitro_, asciminib is 97% bound to plasma proteins, although the specific protein(s) to which it binds are unclear. In mouse tolerability studies, Asciminib dosed BID at increasing concentrations does not cause significant body weight loss, indicating acceptable safety within tested dose ranges [3] |
| References |
[1]. Blood (2014) 124 (21): 398. [2]. Clin Cancer Res (2017) 23 (1_Supplement): IA01. [1]. Nature. 2017, 543: 733-737. |
| Additional Infomation |
Asciminib is a tyrosine kinase inhibitor (TKI) used in the treatment of chronic-phase Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML). More specifically, it is an inhibitor of the ABL1 kinase activity of the BCR-ABL1 fusion protein which serves as a driver of CML proliferation in most patients with the disease. It has also shown benefit in Ph+ CML with the T315I mutation, which produces a mutant BCR-ABL1 which is typically treatment-resistant as compared to wild-type BCR-ABL1. Existing inhibitors of ABL compete at the ATP binding sites of these proteins and can be classified into those that target the active conformation of the kinase domain ([dasatinib], [bosutinib]) and those that target the inactive kinase domain ([imatinib], [nilotinib], [ponatinib]). Asciminib is unique in that it acts as an allosteric inhibitor, binding at the myristoyl pocket of the BCR-ABL1 protein and locking it into an inactive conformation. Asciminib received FDA approval on October 29, 2021 (Scemblix, Novartis AG). Asciminib is a tyrosine kinase inhibitor that specifically targets myristoyl pocket of ABL1 and is used to treat refractory forms of Philadelphia chromosome positive chronic myelocytic leukemia. Serum aminotransferase elevations occur in a proportion of patients treated with asciminib, but episodes of clinically apparent liver injury with jaundice have not been reported with its use. Asciminib is an orally bioavailable, allosteric Bcr-Abl1 tyrosine kinase inhibitor, with antineoplastic activity. Upon administration, asciminib targets and binds to the myristoyl pocket of the Bcr-Abl1 fusion protein at a location that is distinct from the ATP-binding domain, thereby inhibiting the activity of both wild-type Bcr-Abl and certain mutation forms, including the T315I mutation. This binding results in the inhibition of Bcr-Abl1-mediated proliferation and enhanced apoptosis of Philadelphia chromosome-positive (Ph+) hematological malignancies. The Bcr-Abl1 fusion protein tyrosine kinase is an abnormal enzyme produced by leukemia cells that contain the Philadelphia chromosome. See also: Asciminib Hydrochloride (has salt form). Drug Indication Asciminib is indicated for the treatment of adult patients with Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase who have been previously treated with ≥2 tyrosine kinase inhibitors. It is also indicated in the treatment of Ph+ CML in adult patients with the T315I mutation. Scemblix is indicated for the treatment of adult patients with Philadelphia chromosome positive chronic myeloid leukaemia in chronic phase (Ph+ CML CP) previously treated with two or more tyrosine kinase inhibitors (see section 5. 1). Treatment of chronic myeloid leukaemia Mechanism of Action In most patients with chronic myeloid leukemia (CML), progression of the disease is driven primarily by a translocation of the Philadelphia chromosome that creates an oncogenic fusion gene, _BCR-ABL1_, between the _BCR_ and _ABL1_ genes. This fusion gene produces a resultant fusion protein, BCR-ABL1, which exhibits elevated tyrosine kinase and transforming activities that contribute to CML proliferation. Asciminib is an allosteric inhibitor of the BCR-ABL1 tyrosine kinase. It binds to the myristoyl pocket of the ABL1 portion of the fusion protein and locks it into an inactive conformation, preventing its oncogenic activity. Pharmacodynamics Asciminib exerts its therapeutic activity by inhibiting an oncogenic protein responsible for the proliferation of CML. It may be administered orally once or twice a day depending on the condition being treated. By increasing the total daily dose 5-fold as compared to standard therapy (80mg daily vs. 400mg daily), it can be used to treat Ph+ CML with the T315I mutation, a typically treatment-resistant variant of the disease. As with many other chemotherapeutic agents, asciminib treatment can result in various forms of myelosuppression, including thrombocytopenia and neutropenia. Patients should receive frequent laboratory monitoring throughout therapy and dose adjustments may be required based on the severity of observed effects. Patients may also experience pancreatic and/or cardiovascular toxicity, both of which require frequent monitoring and may require dose adjustments as per prescribing information. Asciminib (formerly ABL001) is an allosteric inhibitor undergoing clinical development for chronic myeloid leukemia (CML) and Philadelphia chromosome-positive (Ph⁺) acute lymphoblastic leukemia [3] Its mechanism of action involves binding to the myristoyl pocket of ABL1, distinct from catalytic site inhibitors (imatinib, nilotinib, dasatinib), enabling dual targeting of BCR–ABL1 [3] Pre-existing clonal populations with resistance to Asciminib do not share resistance with nilotinib, supporting combination therapy to block resistance development [2][3] Asciminib has entered phase I clinical testing, showing safety and promising single-agent activity in patients with CML who failed prior tyrosine kinase inhibitor (TKI) therapy [3] In patients with CML achieving deep molecular responses with TKIs, treatment withdrawal can lead to treatment-free remission; Asciminib aims to improve outcomes by eradicating CML cells [2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.56 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.56 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (5.56 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2230 mL | 11.1151 mL | 22.2301 mL | |
| 5 mM | 0.4446 mL | 2.2230 mL | 4.4460 mL | |
| 10 mM | 0.2223 mL | 1.1115 mL | 2.2230 mL |