Physicochemical Properties
| Molecular Formula | C19H20N2O7 |
| Molecular Weight | 388.38 |
| Exact Mass | 388.127 |
| CAS # | 86780-90-7 |
| PubChem CID | 2225 |
| Appearance | Light yellow to yellow solid powder |
| Density | 1.284 g/cm3 |
| Boiling Point | 530ºC at 760 mmHg |
| Melting Point | 155° |
| Flash Point | 274.3ºC |
| Index of Refraction | 1.555 |
| LogP | 2.986 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 8 |
| Rotatable Bond Count | 7 |
| Heavy Atom Count | 28 |
| Complexity | 748 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | NCUCGYYHUFIYNU-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C19H20N2O7/c1-10(22)9-28-19(24)16-12(3)20-11(2)15(18(23)27-4)17(16)13-7-5-6-8-14(13)21(25)26/h5-8,17,20H,9H2,1-4H3 |
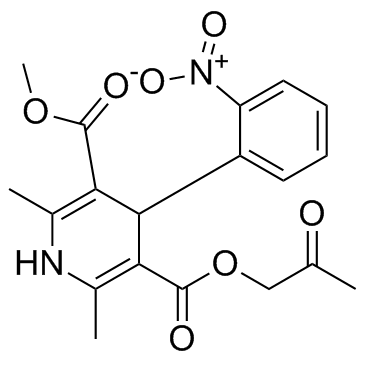
| Chemical Name | 3-O-methyl 5-O-(2-oxopropyl) 2,6-dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate |
| Synonyms | Aranidipine MPC 1304 MPC-1304 MPC1304CCRIS 6724 CCRIS-6724 CCRIS6724 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: This product is not stable in solution, please use freshly prepared working solution for optimal results. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| ln Vivo | A novel Ca2+ channel antagonist for spontaneously hypertensive rats is aranidipine (MPC-1304). Upon administering oral doses of 3 and 10 mg/kg of aradipine to spontaneously hypertensive rats (SHR), there was a significant increase in the Bmax value of specific [3H](+)-PN 200-110 binding to the myocardium in comparison to the control group. to reduce the values. In comparison to the control values, the Bmax values at 1 hour (3 mg/kg), 1 hour, and 6 hours (10 mg/kg) were significantly lower (47.7%, 48.9%, and 25.8%, respectively). The effect peaks after an hour and gradually wanes. The Bmax values following oral administration of alandipine at 6 hours (3 mg/kg) and 12 or 24 hours (10 mg/kg) did not show a significant difference from the control values, suggesting that the alandipine effect was no longer present. The myocardial [3H](+)-PN 200-110 binding's Kd value is unaffected by alandipine taken orally [1]. |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion After administration, aranidipine is rapidly absorbed from the gastrointestinal tract. After absorption, the AUC and Cmax increased linearly in a dose-dependent manner, the Cmax was attained in approximate 3.8-4.8 hours for aranidipine and 4.8-6 hours for the metabolite M-1. The bioavailability of aranidipine in rat, dog, and monkey was about 48%, 41% and 3% respectively. Unchanged aranidipine is found in plasma but not in the urine after 1 hour of administration. Just a small amount of drug was found in the bile. These results indicate that the excretion profile of aranidipine is mainly driven by metabolism and not by excretion. When including the metabolites, 52-56% of the original dose is disposed in the urine, 34-45% in feces and 3-4% in expired air. The excretion in the bile was 59% of the administered dose and 63% of this portion is reabsorbed. Metabolism / Metabolites Eight metabolites of aranidipine were found after oral administration. These metabolites were brought by a reduction of the ketone group, oxidation of dihydropyridine ring and de-esterification at the C-3 position. Biological Half-Life The elimination half-life of aranidipine and the M-1 metabolite are 1.1-1.2 hour and 2.7-3.5 hour respectively. |
| Toxicity/Toxicokinetics |
Protein Binding The binding ratio of plasma proteins of aranidipine varies from 84-95%. This ratio of the drug is similar to the unchanged form and for the M-1 metabolite. Most of the binding happens towards serum albumin and a lower amount corresponds to the alpha1-acid glycoprotein. |
| References |
[1]. Receptor occupation and pharmacokinetics of MPC-1304, a new Ca2+ channel antagonist, in spontaneously hypertensive rats. Eur J Pharmacol. 1995 Dec 12;287(2):191-6. |
| Additional Infomation |
Aranidipine is an organic molecular entity. Aranidipine is a novel dihydropyridine derivative that gives rise to two active metabolites (M-1α and M-1β) that exhibit hypotensive activity. It is a calcium antagonist with the formula methyl 2-oxopropyl 1,4-dihydro-2,6-dimethyl-4-(2-nitrophenyl)-3,5-pyridinedicarboxylate. It was developed by Maruko Seiyaku, introduced by Taiho and launched in Japan in 1997. Drug Indication Aranidipine has been used for many years to treat angina pectoris and hypertension. Mechanism of Action The high potential of aranidipine is thought to be related to the additional calcium antagonistic activity of its metabolite. The mechanism is thought to be related to the capacity of aranidipine and its metabolites to vasodilate afferent and efferent arterioles. this action is performed through the inhibition of voltage-dependent calcium channels. The typical mechanism of action of aranidipine, as all dihydropyridines, is based on the inhibition of L-type calcium channels, decreasing calcium concentration and inducing smooth muscle relaxation. It is a selective alpha2-adrenoreceptor antagonist which inhibits vasoconstrictive responses. Pharmacodynamics Pre-clinical studies with aranidipine and its two metabolites have shown production of increases in femoral blood flow. It has been shown to present potent and long-lasting vasodilating actions. Aranidipine and its metabolites are shown to inhibit calcium-induced contraction in isolated rabbit arteries. Studies have shown that aranidipine is more potent to reduce blood pressure than other dihydropyridines. Aranidipine produce changes in renal blood flow, this effect may be explained by its effect on alpha-2-adrenoreceptor-mediated vasoconstriction. |
Solubility Data
| Solubility (In Vitro) | DMSO : ~125 mg/mL (~321.86 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (5.36 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (5.36 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.08 mg/mL (5.36 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.5748 mL | 12.8740 mL | 25.7480 mL | |
| 5 mM | 0.5150 mL | 2.5748 mL | 5.1496 mL | |
| 10 mM | 0.2575 mL | 1.2874 mL | 2.5748 mL |