Amifampridine (trade name: Firdapse; pyridine-3,4-diamine, 3,4-diaminopyridine, 3,4-DAP) is an FDA approved drug used predominantly in the treatment of a number of rare muscle diseases such as Lambert-Eaton myasthenic syndrome (LEMS) in adults. It was developed by Catalyst pharmaceuticals and gained US approval in November 2018). The free base form of the drug has been used to treat congenital myasthenic syndromes and Lambert–Eaton myasthenic syndrome (LEMS) through compassionate use programs since the 1990s and was recommended as a first line treatment for LEMS in 2006, using ad hoc forms of the drug, since there was no marketed form.
Physicochemical Properties
| Molecular Formula | C5H7N3 |
| Molecular Weight | 109.13 |
| Exact Mass | 109.063 |
| CAS # | 54-96-6 |
| Related CAS # | Amifampridine phosphate;446254-47-3 |
| PubChem CID | 5918 |
| Appearance | Off-white to gray solid powder |
| Density | 1.3±0.1 g/cm3 |
| Boiling Point | 369.3±22.0 °C at 760 mmHg |
| Melting Point | 216-218 °C(lit.) |
| Flash Point | 204.9±9.5 °C |
| Vapour Pressure | 0.0±0.8 mmHg at 25°C |
| Index of Refraction | 1.676 |
| LogP | -0.09 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 3 |
| Rotatable Bond Count | 0 |
| Heavy Atom Count | 8 |
| Complexity | 74.1 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | OYTKINVCDFNREN-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C5H7N3/c6-4-1-2-8-3-5(4)7/h1-3H,7H2,(H2,6,8) |
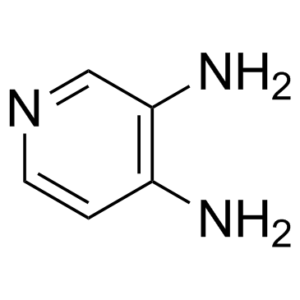
| Chemical Name | 3,4-Diaminopyridine |
| Synonyms | 3,6-DAP; 3,4-Diaminopyridine; BRN-0110232; BRN 0110232; BRN0110232; NSC 521760; NSC-521760; NSC521760; SC10; Trade name: Firdapse. |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | Cav2.1 and Cav1.2 currents in HEK293T cells are unaffected by amifampridine (1.5 μM), whereas Kv3.3 and Kv3.4 currents are severely reduced by about 10% [3]. In frogs and humans, amifampridine (0-100 μM) dose-dependently lengthens the presynaptic AP (action potential) waveforms at the NMJ [3]. |
| ln Vitro |
Cav2.1 and Cav1.2 currents in HEK293T cells are unaffected by amifampridine (1.5 μM), whereas Kv3.3 and Kv3.4 currents are severely reduced by about 10% [3]. In frogs and humans, amifampridine (0-100 μM) dose-dependently lengthens the presynaptic AP (action potential) waveforms at the NMJ [3]. The 3,4-DAP release profile from Lycopodium clavatum exine microcapsules (LEMs) co-encapsulated with shellac was studied in simulated biological fluids. When incubated in simulated gastric fluid (SGF, pH 1.5) for up to 8 hours, the LEMs retained 72.2 ± 4.0% of the loaded 3,4-DAP, demonstrating limited release in an acidic environment. Conversely, substantial 3,4-DAP release was observed upon incubation in phosphate-buffered saline (PBS, pH 7.4), confirming pH-dependent release suitable for enteric delivery. [2] The release was also shown to be time-dependent and proportional to the loading percentage (w/w) of 3,4-DAP (tested at 2.3%, 4.9%, 7.4%, and 10.1%) when measured in PBS (pH 7.4). [2] |
| ln Vivo |
After being intoxicated with BONT/A, amifampridine (10 mg/kg; once) can counteract muscular paralysis [2]. Amifampridine has an hour-long plasma half-life and a roughly 57% bioavailability (F) in mice when administered once at doses of 2.5 mg/kg (IV) and 10 mg/kg (PO) [2]. Amifampridine has a relatively short plasma half-life and, after crossing the blood-brain barrier, can cause epileptic seizures at high concentrations [2]. Four randomised controlled trials involving a total of 54 patients with LEMS showed that 3,4-diaminopyridine treatment resulted in significant improvement in muscle strength score, myometric limb measurement, or compound muscle action potential (CMAP) amplitude. [1] |
| Cell Assay |
Whole-cell patch-clamp electrophysiology in HEK293T cells: HEK293T cells were transfected with plasmids encoding Kv3.3, Kv3.4, Cav2.1, or Cav1.2 channel subunits along with a GFP marker. Recordings were performed at room temperature using an amphotericin-B-based perforated patch configuration. The pipette solution for potassium current recordings contained potassium methane sulfonate, KCl, Hepes, and MgCl2. The bath solution contained NaCl, Hepes, glucose, CaCl2, and MgCl2. For calcium current recordings, cesium-based solutions were used. Cells were voltage-clamped, and currents were activated by depolarizing steps from -100 mV to +20 or +40 mV. Currents were recorded before and after application of 3,4-DAP dissolved in extracellular saline. The percent inhibition was calculated by comparing peak current amplitudes. [3] |
| Animal Protocol |
Animal/Disease Models: CD-1 mice (female, 25 g, 6 weeks old) [2] Doses: 10 mg/kg Route of Administration: BoNT/A administration (IP) followed by po (oral gavage) once (IP) Experimental Results: demonstrated that either LEM alone (182 ± 43 minutes) or the maximum safe oral dose of 3,4-DAP alone (225 ± 24 minutes) Dramatically increased the time to death after toxin administration (216 ± 29 minutes). However, when the 10/50/40 3,4-DAP/LEM/shellac formulation was administered at 25 mg/kg, the time to death was 302 ± 26 minutes, a 40% increase compared to toxin alone. Animal/Disease Models: CD-1 mice (30-35 g, 8 weeks old) [2] Doses: 2.5 mg/kg (IV); 10 mg/kg (PO) Route of Administration: intravenous (iv) (iv)injection, oral administration, once (drug pharmacokinetic/PK/PK analysis) Experimental Results: pharmacokinetic/PK/PK parameters of Amifampridine in CD-1 mice [1]. IV (2.5 mg/kg) PO (10 mg/kg) t1/2 (h) 1.04 1.28 AUC0-24 (μM·h) 4.29 9.72 F (%) 100 56.7 Pharmacokinetic studies were conducted in CD-1 mice. For intravenous (IV) pharmacokinetics, 3,4-DAP was administered intravenously, and plasma concentrations were measured over time. [2] For oral pharmacokinetics and efficacy studies, several formulations were tested: 1) 3,4-DAP phosphate (the clinical salt form) administered orally. 2) LEM formulations with varying compositions: a) LEMs loaded with 7.4% 3,4-DAP and 40% shellac, b) LEMs loaded with 10.1% 3,4-DAP and 40% shellac, c) LEMs loaded with 10.1% 3,4-DAP, 50% LEM, and 40% shellac (optimized ratio). 3) Control formulations: 3,4-DAP mixed with shellac alone (without LEMs), and 3,4-DAP loaded into LEMs without shellac. All oral doses were administered at 25 mg/kg (based on 3,4-DAP) unless specified otherwise for the free drug control (10 mg/kg). [2] For the BoNT/A lethality assay, female CD-1 mice were intoxicated with 5 LD50 of BoNT/A. Treatment groups received either empty LEMs (25 mg/kg LEMs), free 3,4-DAP (10 mg/kg), or the optimized LEM formulation (10.1% 3,4-DAP / 50% LEM / 40% shellac, delivering 25 mg/kg of LEMs per mouse). The primary endpoint was time to death. [2] |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion Orally-administered amifampridine is rapidly absorbed in humans to reach the peak plasma concentrations within by 0.6 to 1.3 hours. A single oral dose of 20 mg amifampridine in fasted individuals resulted in mean peak plasma concentrations (Cmax) ranging from 16 to 137 ng/mL. Bioavailability is approximately 93-100% based on recoveries of unmetabolised amifampridine and a major 3-N-acetylated amifampridine metabolite in urine. Food consumption decreases amifampridine absorption and exposure with a decrease in the time to reach maximum concentrations (Tmax). It is approximated that food consumption lowers the Cmax on average by ~44% and lowers AUC by ~20%. based on geometric mean ratios. Systemic exposure to amifampridine is affected by the overall metabolic acetylation activity of NAT enzymes and NAT2 genotype. The NAT enzymes are highly polymorphic that results in variable slow acetylator (SA) and rapid acetylator (RA) phenotypes. Slow acetylators are more prone to increased systemic exposure to amifampridine, and may require higher doses for therapeutic efficacy. Following oral administration, more than 93% of total amifampridine is renally eliminated within 24 hours. About 19% of the total renally-excreted dose is in the parent drug form, and about 74-81.7% of the dose is in its metabolite form. In healthy volunteers, the volume of distribution for plasma amifampridine indicated that RUZURGI is a drug with a moderate to a high volume of distribution. After a 2 mg/kg infusion in rats, the volume of distribution at steady-state was 2.8 ± 0.7 L/kg. Drug concentrations were highest in organs of excretion, including the liver, kidney, and the gastrointestinal tract, and some tissues of glandular function, such as lacrimal, salivary, mucous, pituitary, and thyroid glands. Concentrations in tissues are generally similar to or greater than concentrations in plasma. Overall clearance of amifampridine is both metabolic and renal; it is primarily cleared from the plasma via metabolism by N-acetylation. Following oral administration of a single 20 or 30 mg dose of RUZURGI to healthy volunteers, amifampridine apparent oral clearance (CL/F) was 149 to 214 L/h. Metabolism / Metabolites Amifampridine is extensively metabolized by N-acetyltransferase 2 (NAT2) to 3-N-acetyl-amifampridine, which is considered an inactive metabolite. Biological Half-Life The average elimination half-life of amifampridine was 3.6 to 4.2 hours and 4.1 to 4.8 hours for the 3-N-acetyl amifampridine metabolite. Following intravenous administration in mice, 3,4-DAP had a plasma half-life of approximately 1 hour. [2] The oral bioavailability (F) of 3,4-DAP phosphate (the immediate-release clinical formulation) in mice was about 57%. This formulation resulted in a rapid spike in plasma concentration. [2] An initial LEM formulation (7.4% 3,4-DAP loading) showed a much lower oral bioavailability of about 17%, but produced a broader and right-shifted plasma concentration peak compared to the immediate-release phosphate salt. [2] Increasing the 3,4-DAP loading in LEMs to 10.1% (with 40% shellac) increased the bioavailability. Further optimization by adjusting the LEM:shellac ratio to 50%:40% (for the 10.1% 3,4-DAP load) dramatically increased the maximum plasma concentration (Cmax) and area under the curve (AUC). The absolute bioavailability of this optimized formulation could not be accurately calculated but was substantially higher. [2] A formulation of 3,4-DAP in shellac alone (without LEMs) failed to generate any measurable plasma concentration. [2] Using the optimized LEM formulation, plasma concentrations of 3,4-DAP remained above the therapeutic threshold of 10-15 µM for over three hours after oral administration. [2] |
| Toxicity/Toxicokinetics |
Hepatotoxicity Amifampridine has had limited clinical use, but adverse events have been largely neurologic and gastrointestinal. Serum ALT elevations were not reported in the prelicensure studies of amifampridine but were reported as occurring in a small proportion of patients in safety reviews by the Food and Drug Administration. Nevertheless, there have been no reports of clinically apparent liver injury associated with its use. Thus, liver injury from amifampridine must be rare if it occurs at all. Likelihood score: E (unlikely cause of clinically apparent liver injury, but experience with its use is limited). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No information is available on the clinical use of amifampridine during breastfeeding on the presence of amifampridine or the 3-N-acetyl-amifampridine metabolite in breastmilk. If amifampridine is required by the mother, it is not a reason to discontinue breastfeeding, but the infant should be carefully monitored for excessive crying or fussiness, adequate weight gain, and developmental milestones. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding In vitro human plasma protein binding of amifampridine and 3-N-acetyl amifampridine was 25.3% and 43.3%, respectively. The most common side effects of 3,4-diaminopyridine are perioral tingling and digital paresthesias; some patients report gastrointestinal symptoms. [1] The most frequent serious adverse event is seizures; this risk appears dose-dependent and has been described at doses around 100 mg per day. [1] Supraventricular tachycardia was reported after iatrogenic intoxication with 360 mg of 3,4-diaminopyridine. [1] One patient died from a myocardial infarction a few weeks after starting the drug, but a causal relationship was unclear. [1] Prolongation of the QT interval is often mentioned as a possible side-effect but was not observed in any of 27 patients studied. [1] |
| References |
[1]. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011 Dec;10(12):1098-107. [2]. Lycopodium clavatum exine microcapsules enable safe oral delivery of 3,4-diaminopyridine for treatment of botulinum neurotoxin A intoxication. Chem Commun (Camb). 2016 Mar 18;52(22):4187-90. [3]. A high-affinity, partial antagonist effect of 3,4-diaminopyridine mediates action potential broadening and enhancement of transmitter release at NMJs. J Biol Chem. 2021 Jan-Jun;296:100302. |
| Additional Infomation |
Pharmacodynamics Administration of amifampridine to patients with LES in clinical trials resulted in improvement of the compound muscle action potential (CMAP), muscle function, and quantitative myasthenia gravis (QMG) score. One case of a slight prolongation of the QTc interval in male patient with LEMS and euthyroid Hashimoto’s disease treated with 90 mg of amifampridine in combination with 100 mg azathioprine was reported. _In vitro_, amifampridine was shown to modulate cardiac conduction and induce phasic contractions in different arteries from several species. In addition, it stimulated potassium-evoked dopamine and noradrenaline release in rat hippocampal slices and upregulate acetylcholine release in the brain. It may also potentiate adrenergic and cholinergic neuromuscular transmission in the gatrointestinal tract. In a single pharmacokinetic study, no effect was observed of amifampridine phosphate on cardiac repolarization as assessed using the QTc interval. There were no changes in heart rate, atrioventricular conduction or cardiac depolarization as measured by the heart rate, PR and QRS interval durations. 3,4-diaminopyridine is the first-choice symptomatic treatment for Lambert-Eaton myasthenic syndrome (LEMS). [1] It is typically administered at 10-20 mg, 2-4 times per day. [1] Its mechanism of action involves blocking potassium channels to prolong depolarization at motor nerve terminals, thereby keeping pathologically affected calcium channels open longer, increasing calcium influx and acetylcholine release. [1] A recent Cochrane review summarized its efficacy based on four randomized controlled trials. [1] Most side effects are dose-dependent, and the peak dose limits its therapeutic window. [1] Potential improvements, such as slow-release tablets or combination with pyridostigmine, are mentioned but require further study. [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (22.91 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (22.91 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (22.91 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 9.1634 mL | 45.8169 mL | 91.6338 mL | |
| 5 mM | 1.8327 mL | 9.1634 mL | 18.3268 mL | |
| 10 mM | 0.9163 mL | 4.5817 mL | 9.1634 mL |