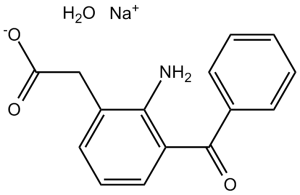
Amfenac Sodium monohydrate (AHR 5850; AHR5850; AHR5850), the sodium salt of amfenac which is an NSAID, is a non-steroidal analgesic anti-inflammatory agent with antipyretic and analgesic activities. It acts as an inhibitor of COX-1 and COX-2 enzymes with IC50s of 15.3 and 20.4 nM, respectively. Amfenac is transformed in vivo to the active metabolite nepafenac which is also a non-steroidal anti-inflammatory drug (NSAID).
Physicochemical Properties
| Molecular Formula | C15H12NO3.H2O.NA | |
| Molecular Weight | 295.27 | |
| Exact Mass | 295.082 | |
| Elemental Analysis | C, 61.02; H, 4.78; N, 4.74; Na, 7.79; O, 21.67 | |
| CAS # | 61618-27-7 | |
| Related CAS # |
|
|
| PubChem CID | 23663941 | |
| Appearance | Typically exists as Light yellow to yellow solid at room temperature | |
| Boiling Point | 520ºC at 760 mmHg | |
| Melting Point | 242-244ºC | |
| Flash Point | 268.3ºC | |
| LogP | 1.309 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 5 | |
| Rotatable Bond Count | 4 | |
| Heavy Atom Count | 21 | |
| Complexity | 342 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | [Na+].O=C(C1C([H])=C([H])C([H])=C([H])C=1[H])C1=C([H])C([H])=C([H])C(C([H])([H])C(=O)[O-])=C1N([H])[H].O([H])[H] |
|
| InChi Key | QZNJPJDUBTYMRS-UHFFFAOYSA-M | |
| InChi Code | InChI=1S/C15H13NO3.Na.H2O/c16-14-11(9-13(17)18)7-4-8-12(14)15(19)10-5-2-1-3-6-10;;/h1-8H,9,16H2,(H,17,18);;1H2/q;+1;/p-1 | |
| Chemical Name | Benzeneacetic acid, 2-amino-3-benzoyl-, sodium salt, monohydrate | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment, avoid exposure to moisture. |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
COX-2; non-steroidal anti-inflammatory drug/NSAIDs Cyclooxygenase-2 (COX-2) (no IC50/Ki; 50 μM Amfenac Sodium Monohydrate inhibited COX-2-mediated prostaglandin E2 (PGE2) synthesis by 68 ± 5% in human uveal melanoma cells, and reduced COX-2 protein expression by 42 ± 4% via Western blot) [1] - Polymorphonuclear Leukocytes (PMNs) Functional Targets (no Ki/IC50; 100 μg/mL Amfenac Sodium Monohydrate inhibited PMN chemotaxis by 55 ± 4%, lysosomal enzyme (β-glucuronidase) release by 48 ± 3%, and superoxide anion (O₂⁻) production by 52 ± 5% in isolated human PMNs) [2] |
| ln Vitro |
The rate of cell proliferation is higher in transfected COX-2-expressing cells than non-transfected ones. All cell lines exhibit a considerable reduction in proliferation rate upon the addition of Amfenac Sodium Hydrate. The addition of melanoma conditioned media inhibits the generation of nitric oxide by macrophages; this inhibition is somewhat alleviated by the addition of amfenac sodium hydroxide[1]. The findings indicate that Amfenac Sodium Hydrate prevents B-glucuronidase from being released: in the presence of 10 -8 and 10-7 M FMLP, respectively, 5×10-4 M Amfenac Sodium Hydrate prevents the release of the enzyme by 35.3 and 16.3%, respectively. After 16 minutes of incubation with 10-8 M FMLP, the aggregation of polymorphonuclear leukocytes (PMNs) is inhibited by 28.3% upon the addition of 10-4 M Amfenac Sodium Hydrate[2]. 1. Inhibition of human uveal melanoma cell proliferation and macrophage NO production ([1]): - Uveal melanoma cell proliferation: Human uveal melanoma cells (OCM-1 line) were cultured in RPMI 1640 + 10% FBS, treated with Amfenac Sodium Monohydrate (1 μM, 10 μM, 50 μM) for 72 h. MTT assay showed dose-dependent inhibition: 10 μM reduced proliferation by 22 ± 3%; 50 μM reduced proliferation by 58 ± 4% vs. control [1] - COX-2/PGE2 regulation: 50 μM Amfenac Sodium Monohydrate reduced COX-2 mRNA expression by 38 ± 3% (RT-PCR) and PGE2 secretion by 68 ± 5% (ELISA) in OCM-1 cells [1] - Macrophage NO production: Human peripheral blood macrophages were stimulated with LPS (1 μg/mL) + IFN-γ (10 ng/mL) to induce NO production, then treated with Amfenac Sodium Monohydrate (50 μM). NO concentration (measured by Griess reagent) was reduced by 45 ± 4% vs. stimulated control [1] 2. Modulation of human PMN functions ([2]): - Chemotaxis inhibition: Isolated human PMNs were placed in the upper chamber of Boyden chambers, with fMLP (10⁻⁷ M) as chemoattractant in the lower chamber. Amfenac Sodium Monohydrate (1 μg/mL, 10 μg/mL, 100 μg/mL) was added to both chambers. 100 μg/mL reduced PMN migration across the filter by 55 ± 4% vs. control [2] - Lysosomal enzyme release: PMNs were stimulated with zymosan (1 mg/mL) to induce lysosomal enzyme release. 100 μg/mL Amfenac Sodium Monohydrate reduced β-glucuronidase release (measured by colorimetric assay) by 48 ± 3% [2] - Oxidative burst inhibition: PMNs were stimulated with phorbol myristate acetate (PMA, 100 ng/mL) to induce O₂⁻ production. 100 μg/mL Amfenac Sodium Monohydrate reduced O₂⁻ levels (measured by cytochrome c reduction assay) by 52 ± 5% [2] |
| ln Vivo | Amfenac possesses both antipyretic and analgesic properties in vivo. Amfenac (4 mg/kg) suppressed acute (Evans blue-carrageenan pleural effusion) and chronic (adjuvant-induced arthritis) inflammation by 33% and 28%, respectively, which is 16.4 and 22.8 times more potent than phenylbutazone. The analgesic activity of Amfenac is 43 times that of acetylsalicylic acid in the Randall-Selitto assay, and 156 and 56.3 times more potent than phenylbutazone in the acetylcholine-induced abdominal constriction in mice and in the bradykinin-induced nociceptive response in dogs, respectively. Amfenac produces less gastric irritation than acetylsalicylic acid when applied topically to the exposed gastric mucosa of cats or when administered orally to rats and dogs. Upon subchronic oral administration to rats, the therapeutic ratio of Amfenac is twice that of phenylbutazone. |
| Enzyme Assay |
1. COX-2 activity assay (human uveal melanoma cells, [1]): - Sample preparation: Human OCM-1 cells were treated with Amfenac Sodium Monohydrate (50 μM) for 24 h, then lysed with RIPA buffer containing protease inhibitors. COX-2-rich lysate (10 μg protein) was collected by centrifugation (12,000×g, 10 min, 4°C). - Reaction system (200 μL): 50 mM Tris-HCl (pH 8.0), 10 mM CaCl₂, 100 μM arachidonic acid (substrate), and COX-2 lysate. - Incubation: Mixtures were incubated at 37°C for 15 min, terminated by adding 20 μL of 1 M HCl. - Detection: PGE2 (COX-2 product) was extracted with ethyl acetate, dried under nitrogen, and measured via ELISA kit. Inhibition rate = (1 - sample PGE2/control PGE2) × 100% [1] |
| Cell Assay |
Human uveal melanoma cell lines were transfected to constitutively express COX-2 and the proliferative rate of these cells using two different methods, with and without the addition of Amfenac, was measured. Nitric oxide production by macrophages was measured after exposure to melanoma-conditioned medium from both groups of cells as well as with and without Amfenac, the active metabolite of Nepafenac. Results: Cells transfected to express COX-2 had a higher proliferation rate than those that did not. The addition of Amfenac significantly decreased the proliferation rate of all cell lines. Nitric oxide production by macrophages was inhibited by the addition of melanoma conditioned medium, the addition of Amfenac partially overcame this inhibition. Conclusion: Amfenac affected both COX-2 transfected and non-transfected uveal melanoma cells in terms of their proliferation rates as well as their suppressive effects on macrophage cytotoxic activity[1]. 1. Human uveal melanoma cell proliferation and COX-2 assay ([1]): - Cell culture: OCM-1 cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37°C in 5% CO₂. - Proliferation assay: Cells were plated in 96-well plates (5×10³ cells/well), adhered overnight, then treated with Amfenac Sodium Monohydrate (1 μM, 10 μM, 50 μM) for 72 h. MTT (5 mg/mL) was added for 4 h, DMSO dissolved formazan, and absorbance at 570 nm was measured. Proliferation inhibition rate = (1 - sample absorbance/control absorbance) × 100% [1] - COX-2 protein detection: Cells were lysed with RIPA buffer, 30 μg protein was separated by 10% SDS-PAGE, transferred to PVDF membrane. Membrane was incubated with anti-COX-2 primary antibody and HRP-conjugated secondary antibody, visualized by ECL. Band intensity was quantified by ImageJ, normalized to GAPDH [1] 2. Human PMN isolation and function assay ([2]): - PMN isolation: Human venous blood was mixed with 3.8% sodium citrate (1:9 v/v), layered on Ficoll-Hypaque gradient, centrifuged (400×g, 30 min, 20°C). PMN-rich pellet was collected, treated with hypotonic buffer to lyse red blood cells, washed with PBS, resuspended in HBSS (1×10⁷ cells/mL). - Chemotaxis assay: Boyden chambers with 8 μm pore filter were used. Upper chamber: PMNs + Amfenac Sodium Monohydrate (1-100 μg/mL); lower chamber: HBSS + fMLP (10⁻⁷ M). Incubated at 37°C for 90 min, filter was stained, migrated PMNs in 5 high-power fields were counted [2] - Lysosomal enzyme release assay: PMNs (1×10⁶ cells/mL) + Amfenac Sodium Monohydrate (1-100 μg/mL) were incubated for 30 min, then zymosan (1 mg/mL) was added for 1 h. Supernatant was collected, β-glucuronidase activity was measured by colorimetric assay (405 nm absorbance with p-nitrophenyl β-D-glucuronide as substrate) [2] |
| Animal Protocol |
topical Cats, rats, dogs |
| Toxicity/Toxicokinetics |
rat LD50 oral 311 mg/kg BEHAVIORAL: CHANGES IN MOTOR ACTIVITY (SPECIFIC ASSAY); BEHAVIORAL: ATAXIA; GASTROINTESTINAL: OTHER CHANGES Agents and Actions, A Swiss Journal of Pharmacology., 7(133), 1977 [PMID:871089] rat LD50 subcutaneous 240 mg/kg Iyakuhin Kenkyu. Study of Medical Supplies., 16(1461), 1985 rat LD50 intravenous 277 mg/kg BEHAVIORAL: SOMNOLENCE (GENERAL DEPRESSED ACTIVITY); GASTROINTESTINAL: HYPERMOTILITY, DIARRHEA Journal of Toxicological Sciences., 9(87), 1984 [PMID:6471132] rat LD50 intramuscular 277 mg/kg Iyakuhin Kenkyu. Study of Medical Supplies., 16(1461), 1985 mouse LD50 oral 615 mg/kg BEHAVIORAL: CHANGES IN MOTOR ACTIVITY (SPECIFIC ASSAY); GASTROINTESTINAL: ULCERATION OR BLEEDING FROM STOMACH; BLOOD: OTHER CHANGES Agents and Actions, A Swiss Journal of Pharmacology., 7(133), 1977 [PMID:871089] 1. In vitro safety: - Amfenac Sodium Monohydrate at concentrations up to 50 μM had no significant cytotoxicity on normal human retinal pigment epithelial (RPE) cells (MTT assay: viability ≥85% vs. control) after 72 h treatment [1] - At 100 μg/mL, Amfenac Sodium Monohydrate did not induce PMN necrosis (trypan blue exclusion assay: viability ≥90% vs. control) [2] |
| References |
[1]. Marshall JC, et al. The effects of a cyclooxygenase-2 (COX-2) expression and inhibition on human uveal melanoma cell proliferation and macrophage nitric oxide production. J Carcinog. 2007 Nov 27;6:17. [2]. Matsumoto T, et al. Effect of a non-steroidal anti-inflammatory drug (amfenac sodium) on polymorphonuclear leukocytes. Pharmacol Res Commun. 1982 Jun;14(6):523-32 |
| Additional Infomation |
Amfenac sodium hydrate is a hydrate that is the monohydrate of the sodium salt of amfenac. It has a role as an antipyretic, a cyclooxygenase 2 inhibitor, a cyclooxygenase 1 inhibitor, a non-narcotic analgesic and a non-steroidal anti-inflammatory drug. It contains a sodium (2-amino-3-benzoylphenyl)acetate. 1. Amfenac Sodium Monohydrate is a non-steroidal anti-inflammatory drug (NSAID) with dual anti-inflammatory mechanisms: (1) inhibiting COX-2 to reduce prostaglandin synthesis (targeting chronic inflammation); (2) suppressing PMN functions (chemotaxis, lysosomal enzyme release, oxidative burst) to alleviate acute inflammatory responses [1,2] 2. In ophthalmology-related research ([1]), Amfenac Sodium Monohydrate inhibits uveal melanoma cell proliferation by downregulating COX-2, suggesting potential adjuvant therapeutic value for ocular malignancies associated with COX-2 overexpression [1] 3. Its inhibitory effect on PMNs ([2]) indicates efficacy in treating acute inflammatory diseases (e.g., bacterial infections, tissue injury) where PMN infiltration and activation play a key role [2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.3867 mL | 16.9337 mL | 33.8673 mL | |
| 5 mM | 0.6773 mL | 3.3867 mL | 6.7735 mL | |
| 10 mM | 0.3387 mL | 1.6934 mL | 3.3867 mL |