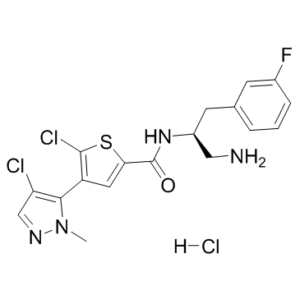
Afuresertib HCl (also named as GSK2110183 HCl) is a potent, orally bioavailable and ATP-competitive Akt inhibitor with Ki of 0.08 nM, 2 nM, and 2.6 nM for Akt1, Akt2, and Akt3, respectively. Afuresertib is a protein kinase B (Akt) serine/threonine inhibitor with potential anti-cancer properties. The PI3K/Akt signaling pathway, tumor cell proliferation, and induction of tumor cell apoptosis may all be inhibited as a result of the Akt inhibitor GSK2110183's binding to and inhibition of Akt activity. The PI3K/Akt signaling pathway is frequently involved in the development of tumors, and dysregulated PI3K/Akt signaling may be a factor in the tumors' resistance to a number of different anti-cancer drugs.
Physicochemical Properties
| Molecular Formula | C₁₈H₁₈CL₃FN₄OS |
| Molecular Weight | 463.78 |
| Exact Mass | 462.025 |
| Elemental Analysis | C, 46.62; H, 3.91; Cl, 22.93; F, 4.10; N, 12.08; O, 3.45; S, 6.91 |
| CAS # | 1047645-82-8 |
| Related CAS # | Afuresertib;1047644-62-1; 1047645-82-8 (HCl); 1047634-63-8 (Afuresertib-F free base); 2070009-64-0 (Afuresertib-F HCl) |
| PubChem CID | 46843056 |
| Appearance | White to off-white solid powder |
| LogP | 5.787 |
| Hydrogen Bond Donor Count | 3 |
| Hydrogen Bond Acceptor Count | 5 |
| Rotatable Bond Count | 6 |
| Heavy Atom Count | 28 |
| Complexity | 520 |
| Defined Atom Stereocenter Count | 1 |
| SMILES | ClC1=C(C2=C(C([H])=NN2C([H])([H])[H])Cl)C([H])=C(C(N([H])[C@]([H])(C([H])([H])N([H])[H])C([H])([H])C2C([H])=C([H])C([H])=C(C=2[H])F)=O)S1.Cl[H] |
| InChi Key | YFQJOPFTGMHYNV-YDALLXLXSA-N |
| InChi Code | InChI=1S/C18H17Cl2FN4OS.ClH/c1-25-16(14(19)9-23-25)13-7-15(27-17(13)20)18(26)24-12(8-22)6-10-3-2-4-11(21)5-10;/h2-5,7,9,12H,6,8,22H2,1H3,(H,24,26);1H/t12-;/m0./s |
| Chemical Name | N-[(2S)-1-amino-3-(3-fluorophenyl)propan-2-yl]-5-chloro-4-(4-chloro-2-methylpyrazol-3-yl)thiophene-2-carboxamide;hydrochloride |
| Synonyms | GSK2110183; GSK 2110183; ASB183; ASB-183; GSK 2110183C; Afuresertib (GSK2110183); GSK-2110183; GSK2110183B; GSK2110183B; GSK 2110183B; 1047645-82-8; GSK2110183B; Afuresertib hydrochloride [USAN]; UNII-0FC27E442O; 0FC27E442O; Afuresertib HCl; Afuresertib hydrochloride |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: (1). This product requires protection from light (avoid light exposure) during transportation and storage.(2). Please store this product in a sealed and protected environment (e.g. under nitrogen), avoid exposure to moisture. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Akt1 (Ki = 0.08 nM); Akt2 (Ki = 2 nM); Akt3 (Ki = 2.6 nM); Akt1 E17K mutant (IC50 = 0.2 nM); PKCη (IC50 = 210 nM); PKC-βI (IC50 = 430 nM); ROCK (IC50 = 100 nM); PKCθ (IC50 = 510 nM) Afuresertib is a catalytic, ATP‑competitive inhibitor targeting AKT1, AKT2, and AKT3. [1] |
| ln Vitro |
Afuresertib (GSK 2110183) exhibits favorable tumor-suppressive effects on malignant pleural mesothelioma (MPM) cells.Afuresertib significantly raises the activity of caspase-3 and caspase-7 as well as the proportion of apoptotic cells in ACC-MESO-4 and MSTO-211H cells. The cell cycle is strongly stopped by afuresertib in the G1 phase. Afuresertib increases the expression of p21WAF1/CIP1 and decreases the phosphorylation of Akt substrates, such as GSK-3 and FOXO family proteins, according to Western blotting analysis. By stimulating FOXO activity, afuresertib-induced p21 expression encourages G1 phase arrest. Afuresertib significantly increases the cytotoxicity that cisplatin causes. Afuresertib alters the expression of the genes MYC and E2F1, which are related to the fibroblast core serum response[1]. Afuresertib suppresses the viability of six malignant pleural mesothelioma (MPM) cell lines (ACC‑MESO‑4, MSTO‑211H, NCI‑H2052, NCI‑H28, NCI‑H290, Y‑MESO‑8A) in a dose‑dependent manner, with IC₅₀ values ranging from 4.2 µM to 12.1 µM. The IC₅₀ values for afuresertib against MPM cell lines are lower than those against the normal mesothelial cell line MeT‑5A (IC₅₀ = 17.8 µM), indicating tumor‑specific antiproliferative activity. Afuresertib (10 µM) significantly inhibits colony formation of ACC‑MESO‑4, MSTO‑211H, and NCI‑H2052 cells. Afuresertib (20 µM) induces apoptosis in ACC‑MESO‑4 and MSTO‑211H cells, as shown by increased Annexin V‑FITC/PI‑positive cells and elevated caspase‑3/7 activities. Afuresertib increases the protein levels of pro‑apoptotic molecules BIM and BAX, and cleaved caspase‑3. Afuresertib (5–10 µM) arrests the cell cycle at G₀/G₁ phase, decreases the population of cells in S and G₂‑M phases, and reduces phosphorylation of Akt substrates (GSK3β, mTOR, p70 S6K) and YAP (Ser127). Afuresertib down‑regulates the expression of E2F1, CDK4, phospho‑CDK2, and up‑regulates p21WAF1/CIP1. Afuresertib decreases phosphorylation of FOXO1 (Thr24, Ser256). Afuresertib (5 µM) inhibits migration of ACC‑MESO‑4 and MSTO‑211H cells in a scratching assay. Afuresertib (20 µM) synergistically enhances cisplatin‑induced cytotoxicity and apoptosis in ACC‑MESO‑4 and MSTO‑211H cells. Afuresertib also enhances pemetrexed‑induced apoptosis in MSTO‑211H cells. Gene‑expression profiling shows that afuresertib down‑regulates oncogenic signature genes related to Akt signaling, serum response, E2F1, MYC, and mTOR pathways.[1] |
| ln Vivo |
GSK2110183 is administered orally to mice bearing BT474 breast tumor xenografts at doses of 10, 30, or 100 mg/kg every day for 21 days, resulting in 8, 37, or 61% TGI, respectively. Mice tolerated GSK2110183 well; after 5 days of dosing, there was a 1-3% loss of body weight, which recovered throughout the course of the study. To further demonstrate the effectiveness of the compound, other tumor xenograft models with activated Akt pathways are investigated. Mice given GSK2110183 at doses of 10, 30, and 100 mg/kg develop SKOV3 xenografts with TGIs of 23, 37, and 97%, respectively[2]. In female SCID mice bearing established BT474 breast tumor xenografts, daily oral administration of afuresertib at 10, 30, and 100 mg/kg for 21 days resulted in tumor growth inhibition (TGI) of 8%, 37%, and 61%, respectively, compared to vehicle control. In female athymic nude mice bearing SKOV3 ovarian tumor xenografts, daily oral administration of afuresertib at 10, 30, and 100 mg/kg for 21 days resulted in TGI of 23%, 37%, and 97%, respectively. In combination studies, afuresertib (100 mg/kg, QD) combined with the MEK inhibitor trametinib (GSK1120212) in HPAC pancreatic cancer xenograft models showed enhanced anti-tumor efficacy compared to either agent alone. Immunohistochemical analysis of HPAC tumors treated with the combination showed reduced proliferation (Ki67), increased apoptosis (cleaved caspase 3), decreased phospho-PRAS40 and phospho-S6 levels, and feedback hyperphosphorylation of AKT. [2] |
| Enzyme Assay |
MPM cells are plated in 96-well plates at a cell density of 2.5 103 per well, and then they are left to grow for 24 hours at 37°C. Following that, the cells are incubated for 72 hours in a medium containing the Akt inhibitors at the indicated concentrations (e.g., Afuresertib; 50, 20, 10, 5, 2, 1, 0.5, 0.2, 0.1, and 0.01 M). The cells are then incubated for 4 hours with MTT solution added to each well. The cells are then given an overnight incubation in lysis buffer (10% SDS in 0.01 mol/L hydrogen chloride). Absorbance is measured at 550 nm using SpectraMAX M5 spectrophotometer[1]. The potency of afuresertib against AKT enzymes was measured using a filter binding assay. A pre-mix of enzyme and inhibitor was incubated for 1 hour, then added to a GSK3α peptide substrate and [γ³³P]ATP. Reactions were terminated after 2 hours, and radiolabeled phosphorylated peptide product was captured on a phospho-cellulose filter plate for quantification. Progress curve analysis was also performed using continuous real-time fluorescence detection of product formation with a Sox-AKT-tide substrate. Afuresertib was tested against a diverse panel of kinase assays (261 kinases) at concentrations of 0.5 µM and 10 µM. Full IC₅₀ curves were generated for kinases showing >50% inhibition at 0.5 µM. [2] |
| Cell Assay |
Apoptosis assay[1] Apoptosis was evaluated by performing AxV–FITC/PI double staining‐based FACS analysis, as described previously 25. Briefly, ACC‐MESO‐4 and MSTO‐211H cells were seeded in six‐well plates (cell density, 1 × 105 cells/well) and were incubated for 24 h at 37°C. Next, the cells were incubated with indicated concentrations of afuresertib, followed by incubation with AxV–FITC and PI (10 μg/mL) for 15 min at room temperature. Fluorescence intensities were determined by performing FACS with FACSCantoII. Cell cycle analysis[1] Cell cycle was evaluated by performing PI‐staining‐based FACS analysis, as described previously 26. ACC‐MESO‐4 and MSTO‐211H cells were seeded in a six‐well culture plate (cell density, 1 × 105 cells/well) and were incubated for 24 h. Next, the cells were incubated with the indicated concentrations of afuresertib for 24 h. For FACS analysis, the cells were detached using trypsin after 24 h of serum treatment and were fixed overnight in ice‐cold 70% ethanol. After fixation, the cells were treated with RNase A (100 μg/mL) and stained with PI (10 μg/mL). The percentages of cells in the sub‐G1, G1, S, and G2‐M phases of the cell cycle were measured using FlowJo software. A 3-day proliferation assay using CellTiter-Glo is performed to measure the growth inhibition by the compounds at 0-30 μM. The rate of cell growth is measured in comparison to untreated (DMSO) controls. In the Assay Client application, EC50 values are calculated from inhibition curves using a 4- or 6-parameter fitting algorithm.[2] For cell viability assay, MPM cells are seeded in 96‑well plates at 2.5×10³ cells/well and incubated for 24 h. Cells are then treated with various concentrations of afuresertib for 72 h. MTT solution is added and incubated for 4 h, followed by overnight incubation with lysis buffer (10% SDS in 0.01 mol/L HCl). Absorbance is measured at 550 nm.[1] For apoptosis assay, cells are seeded in 6‑well plates at 1×10⁵ cells/well, treated with afuresertib for 48 h, then stained with Annexin V‑FITC and propidium iodide (10 µg/mL) for 15 min at room temperature. Fluorescence is measured by flow cytometry.[1] For cell‑cycle analysis, cells are treated with afuresertib for 24 h, fixed in ice‑cold 70% ethanol overnight, treated with RNase A (100 µg/mL), stained with propidium iodide (10 µg/mL), and analyzed by flow cytometry.[1] For colony‑formation assay, cells are seeded at 200 cells/well in 6‑well plates, treated with 10 µM afuresertib for 14 days, then stained with crystal violet and counted.[1] For cell‑confluence proliferation assay, cells are seeded in 12‑well plates at 1×10⁴ cells/well and treated with afuresertib. Phase images are recorded using a live‑cell imaging system.[1] For scratching assay, cells are grown to 80–90% confluence in 24‑well plates, scratched with a pipette tip, and then treated with afuresertib. Migration is monitored by live‑cell imaging.[1] For quantitative RT‑PCR, total RNA is extracted after afuresertib treatment, reverse transcribed, and analyzed using TaqMan probes.[1] For Western blotting, cells are lysed in loading buffer after afuresertib treatment, and proteins are detected using specific antibodies.[1] |
| Animal Protocol |
Dissolved in 20% polyethylene glycol (PEG) 400/1% DMSO; 25, 50, 100 mg/kg; p.o.Female athymic nude and SCID mice bearing SKOV3 or BT474 tumors
Female athymic nude and SCID mice bearing SKOV3 or BT474 tumors[1] 100 mg/kg p.o. In vivo Xenograft experiments[2] Tumors were initiated by injecting either cells (SKOV3, CAPAN-2 and HPAC) or a tumor fragments (BT474) subcutaneously into 6–8 week female athymic nude (SKOV3) and SCID (all others) mice. Once tumors reached between 120 and 300 mm3, mice were randomized according to tumor volume into groups of n = 7–10 mice per treatment. GSK2110183 and GSK2141795 were administered daily at various doses by oral gavage. In combination experiments, GSK1120212 was also administered daily by oral gavage. Tumor volumes and body weight were measured twice weekly, tumor volume was measured with calipers and calculated using equation: Tumor volume (mm3) = (length x width)2/2. Results are represented as percent inhibition on completion of dosing = 100 x [1- average growth of drug-treated population/average growth of vehicle-treated control population].[2] In vivo dose response pharmacodynamic assay[2] SCID mice bearing BT474 tumor xenografts were treated with either vehicle, GSK2110183 or GSK2141795 daily for 7 days prior to harvesting tissue 2 h post the last dose. Protein lysates were analyzed by phospho-PRAS40 ELISA according to the methods described above. Concentration of the test compounds in the tissue and blood was analyzed using protein precipitation with acetonitrile, followed by HPLC/MS/MS analysis using positive ion atmospheric pressure chemical ionization or Turbo ionspray ionization. The lower level of detection of compound was 10 ng/mL and the assays were linear over a 100- to a 1000-fold drug concentration range. For in vivo xenograft efficacy studies, tumors were established in female nu/nu CD-1 or SCID mice by subcutaneous injection of cancer cells or tumor fragments. When tumors reached 120–300 mm³, mice were randomized into groups (n=7-10). Afuresertib was formulated in 20% polyethylene glycol (PEG) 400 / 1% DMSO and administered daily by oral gavage at specified doses (e.g., 10, 30, 100 mg/kg) for 21 days. Tumor volumes were measured twice weekly using calipers, and body weight was monitored. For pharmacodynamic studies, mice bearing BT474 tumors were treated with afuresertib daily for 7 days. Tumors and blood were harvested at various time points post-last dose for analysis of phospho-PRAS40 levels by ELISA and compound concentration by LC/MS-MS. For combination studies with the MEK inhibitor trametinib, both drugs were administered daily by oral gavage. Trametinib was formulated in 0.5% hydroxypropylmethylcellulose and 0.2% Tween-80 (pH 8.0). [2] |
| ADME/Pharmacokinetics |
Afuresertib shows high plasma protein binding (>95% in human and rodent plasma). In BT474 tumor-bearing mice, blood concentrations of approximately 3–4 µM (Cmax) result in ~60% inhibition of phospho-PRAS40 in tumors, sustained for 24 hours after a single 100 mg/kg oral dose. Tumor exposure of afuresertib is consistently higher than blood exposure (≥3-fold). A time-course experiment shows that phospho-PRAS40 inhibition returns to baseline by 48 hours post-dose. [2] |
| Toxicity/Toxicokinetics |
Administration of a single 100 mg/kg oral dose of afuresertib to mice results in a transient increase in blood glucose (peak ~211 mg/dL at 2 hours post-dose) and plasma insulin (peak ~105.6 ng/mL at 4 hours post-dose). Levels return to normal by 8 hours post-dose. Minimal body weight loss (1–3%) was observed in mice during 21-day efficacy studies, which recovered over the course of the study. [2] |
| References |
[1]. Novel ATP-competitive Akt inhibitor Afuresertib suppresses the proliferation of malignant pleural mesothelioma cells. Cancer Med. 2017 Nov;6(11):2646-2659. [2]. Discovery of novel AKT inhibitors with enhanced anti-tumor effects in combination with the MEK inhibitor. PLoS One. 2014 Jun 30;9(6):e100880. |
| Additional Infomation |
N-[(2S)-1-amino-3-(3-fluorophenyl)propan-2-yl]-5-chloro-4-(4-chloro-2-methyl-3-pyrazolyl)-2-thiophenecarboxamide is a member of amphetamines.\n \nAfuresertib has been used in trials studying the treatment of Cancer and Neoplasms, Haematologic.\n \nAfuresertib is an orally bioavailable inhibitor of the serine/threonine protein kinase Akt (protein kinase B) with potential antineoplastic activity. Afuresertib binds to and inhibits the activity of Akt, which may result in inhibition of the PI3K/Akt signaling pathway and tumor cell proliferation and the induction of tumor cell apoptosis. Activation of the PI3K/Akt signaling pathway is frequently associated with tumorigenesis and dysregulated PI3K/Akt signaling may contribute to tumor resistance to a variety of antineoplastic agents. \nMalignant pleural mesothelioma (MPM), an asbestos‐related occupational disease, is an aggressive and incurable tumor of the thoracic cavity. Despite recent advances in MPM treatment, overall survival of patients with MPM is very low. Recent studies have implicated that PI3K/Akt signaling is involved in MPM cell survival and development. To investigate the effects of Akt inhibitors on MPM cell survival, we examined the effects of nine selective Akt inhibitors, namely, afuresertib, Akti‐1/2, AZD5363, GSK690693, ipatasertib, MK‐2206, perifosine, PHT‐427, and TIC10, on six MPM cell lines, namely, ACC‐MESO‐4, Y‐MESO‐8A, MSTO‐211H, NCI‐H28, NCI‐H290, and NCI‐H2052, and a normal mesothelial cell line MeT‐5A. Comparison of IC 50 values of the Akt inhibitors showed that afuresertib, an ATP‐competitive specific Akt inhibitor, exerted tumor‐specific effects on MPM cells. Afuresertib significantly increased caspase‐3 and caspase‐7 activities and apoptotic cell number among ACC‐MESO‐4 and MSTO‐211H cells. Moreover, afuresertib strongly arrested the cell cycle in the G1 phase. Western blotting analysis showed that afuresertib increased the expression of p21WAF 1/ CIP 1 and decreased the phosphorylation of Akt substrates, including GSK‐3β and FOXO family proteins. These results suggest that afuresertib‐induced p21 expression promotes G1 phase arrest by inducing FOXO activity. Furthermore, afuresertib significantly enhanced cisplatin‐induced cytotoxicity. Interestingly, results of gene set enrichment analysis showed that afuresertib modulated the expression E2F1 and MYC, which are associated with fibroblast core serum response. Together, these results suggest that afuresertib is a useful anticancer drug for treating patients with MPM.[1] \n\n\nTumor cells upregulate many cell signaling pathways, with AKT being one of the key kinases to be activated in a variety of malignancies. GSK2110183 and GSK2141795 are orally bioavailable, potent inhibitors of the AKT kinases that have progressed to human clinical studies. Both compounds are selective, ATP-competitive inhibitors of AKT 1, 2 and 3. Cells treated with either compound show decreased phosphorylation of several substrates downstream of AKT. Both compounds have desirable pharmaceutical properties and daily oral dosing results in a sustained inhibition of AKT activity as well as inhibition of tumor growth in several mouse tumor models of various histologic origins. Improved kinase selectivity was associated with reduced effects on glucose homeostasis as compared to previously reported ATP-competitive AKT kinase inhibitors. In a diverse cell line proliferation screen, AKT inhibitors showed increased potency in cell lines with an activated AKT pathway (via PI3K/PTEN mutation or loss) while cell lines with activating mutations in the MAPK pathway (KRAS/BRAF) were less sensitive to AKT inhibition. Further investigation in mouse models of KRAS driven pancreatic cancer confirmed that combining the AKT inhibitor, GSK2141795 with a MEK inhibitor (GSK2110212; trametinib) resulted in an enhanced anti-tumor effect accompanied with greater reduction in phospho-S6 levels. Taken together these results support clinical evaluation of the AKT inhibitors in cancer, especially in combination with MEK inhibitor.[2] Afuresertib is an ATP‑competitive Akt inhibitor that shows promising tumor‑suppressive effects in MPM cells by inhibiting Akt signaling, inducing apoptosis, arresting cell cycle, and enhancing chemotherapy efficacy. The study suggests that afuresertib could be a potential therapeutic agent for malignant pleural mesothelioma, especially in combination with cisplatin.[1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.39 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.39 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (5.39 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1562 mL | 10.7810 mL | 21.5619 mL | |
| 5 mM | 0.4312 mL | 2.1562 mL | 4.3124 mL | |
| 10 mM | 0.2156 mL | 1.0781 mL | 2.1562 mL |