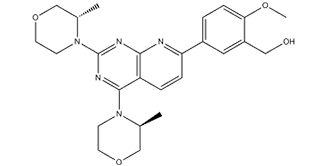
AZD8055 is a novel, potent selective, and orally bioavailable ATP-competitive mTOR (mammalian target of rapamycin) inhibitor with potential anticancer activity. It has excellent selectivity (∼1,000-fold) against PI3K isoforms and ATM/DNA-PK and inhibits mTOR with an IC50 of 0.8 nM in MDA-MB-468 cells. In its role as an mTOR inhibitor, AZD-8055 demonstrated strong anti-tumor activity by preventing mTOR's serine/threonine kinase activity. This decreased expression of mRNAs required for cell cycle progression may result in cell cycle arrest and tumor cell apoptosis. Transcriptional factors phosphorylated by mTOR include S6K1 and 4E-BP1, which promote protein synthesis and control cell growth, proliferation, motility, and survival.
Physicochemical Properties
| Molecular Formula | C25H31N5O4 | |
| Molecular Weight | 465.5447 | |
| Exact Mass | 465.237 | |
| Elemental Analysis | C, 64.50; H, 6.71; N, 15.04; O, 13.75 | |
| CAS # | 1009298-09-2 | |
| Related CAS # |
|
|
| PubChem CID | 25262965 | |
| Appearance | Yellow solid powder | |
| Density | 1.2±0.1 g/cm3 | |
| Boiling Point | 694.3±65.0 °C at 760 mmHg | |
| Flash Point | 373.7±34.3 °C | |
| Vapour Pressure | 0.0±2.3 mmHg at 25°C | |
| Index of Refraction | 1.609 | |
| LogP | 0.27 | |
| Hydrogen Bond Donor Count | 1 | |
| Hydrogen Bond Acceptor Count | 9 | |
| Rotatable Bond Count | 5 | |
| Heavy Atom Count | 34 | |
| Complexity | 659 | |
| Defined Atom Stereocenter Count | 2 | |
| SMILES | O1C([H])([H])C([H])([H])N(C2C3C([H])=C([H])C(C4C([H])=C([H])C(=C(C([H])([H])O[H])C=4[H])OC([H])([H])[H])=NC=3N=C(N=2)N2C([H])([H])C([H])([H])OC([H])([H])[C@]2([H])C([H])([H])[H])[C@@]([H])(C([H])([H])[H])C1([H])[H] |
|
| InChi Key | KVLFRAWTRWDEDF-IRXDYDNUSA-N | |
| InChi Code | InChI=1S/C25H31N5O4/c1-16-14-33-10-8-29(16)24-20-5-6-21(18-4-7-22(32-3)19(12-18)13-31)26-23(20)27-25(28-24)30-9-11-34-15-17(30)2/h4-7,12,16-17,31H,8-11,13-15H2,1-3H3/t16-,17-/m0/s1 | |
| Chemical Name | [5-[2,4-bis[(3S)-3-methylmorpholin-4-yl]pyrido[2,3-d]pyrimidin-7-yl]-2-methoxyphenyl]methanol | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
mTOR (IC50 = 0.13 nM); mTOR (IC50 = 0.8 nM) AZD8055 is a potent, ATP-competitive inhibitor of mammalian target of rapamycin (mTOR), targeting both mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). In recombinant enzyme assays, it exhibits IC50 values of 0.8 nM for mTORC1 (measured by GST-S6K1 phosphorylation inhibition) and 0.4 nM for mTORC2 (measured by GST-Akt Ser473 phosphorylation inhibition), with minimal activity against class I PI3K subtypes (IC50 > 1000 nM for PI3Kα/β/γ/δ) [1] - In human acute myeloid leukemia (AML) MV4-11 cells (FLT3-mutant), AZD8055 inhibits mTOR-mediated Akt Ser473 phosphorylation with an EC50 of 0.06 μM, without affecting total Akt protein levels [2] - In human pediatric rhabdomyosarcoma RD cells, AZD8055 suppresses mTORC1-mediated S6 ribosomal protein phosphorylation (Ser235/236) with an EC50 of 0.08 μM [3] |
| ln Vitro |
AZD8055 shows low activity (∼1,000-fold) against all PI3K isoforms (α, β, γ, δ) and other members of the PI3K-like kinase family (ATM and DNA-PK). AZD8055 prevents the phosphorylation of mTORC1 (p70S6K and 4E-BP1), mTORC2 (Akt), and downstream proteins. A significant amount of cap-dependent translation can be inhibited by AZD8055 because it completely inhibits the rapamycin-resistant T37/46 phosphorylation sites on 4E-BP1. With IC50 values of 53, 50, and 20 nM, respectively, AZD8055 potently inhibits proliferation in U87MG, A549, and H838 cells. In addition, autophagy and elevated LC3-II levels are induced by AZD8055 in H838 and A549 cells.[1] AML blast cell proliferation and cell cycle progression are reduced, leukemic progenitors' clonogenic growth is inhibited, and AZD8055 induces caspase-dependent apoptosis in leukemic cells but not in healthy, immature CD34+ cells.[2] With an IC50 of 24.7 nM, AZD8055 exhibits inhibitory activity against the pediatric preclinical testing program (PPTP) cell lines and causes appreciable variations in EFS distribution. [3] In human non-small cell lung cancer (NSCLC) A549 cells (PTEN-deficient), AZD8055 (0.01-10 μM) inhibited cell proliferation in a dose-dependent manner, with an IC50 of 0.12 μM after 72 hours (MTT assay). Western blot analysis showed that 1 μM AZD8055 reduced phosphorylation of mTORC1 targets (p-S6 Ser235/236, -90%; p-4E-BP1 Thr37/46, -85%) and mTORC2 target (p-Akt Ser473, -88%) within 24 hours. Flow cytometry (Annexin V-FITC/PI) revealed that 2 μM AZD8055 increased the apoptotic rate from 3% (control) to 40% [1] - In human AML MV4-11 cells, AZD8055 (0.01-2 μM) induced dose-dependent cell death. The IC50 was 0.07 μM after 48 hours (SRB assay). At 0.2 μM, it activated caspase-3 (3.2-fold increase in cleaved caspase-3) and reduced p-mTOR Ser2448 (-90%) and p-FLT3 (-45%) (via feedback inhibition) [2] - In human pediatric rhabdomyosarcoma RD cells and neuroblastoma SK-N-SH cells, AZD8055 (0.05-2 μM) inhibited proliferation with IC50 values of 0.09 μM (RD) and 0.11 μM (SK-N-SH) after 72 hours. At 0.1 μM, it reduced colony formation by 70% (RD cells, 14-day crystal violet staining) and downregulated the stemness marker SOX2 (-65%) [3] |
| ln Vivo |
AZD8055 inhibits the pS6 and pAkt in U87MG and A549 xenografts at 2.5/10 mg/kg, which leads to tumor growth inhibition. At doses of 10–20 mg/kg, AZD8055 significantly inhibits tumor growth in a variety of xenografts, including U87MG, BT474c, A549, Calu-3, LoVo, SW620, PC3, and MES-SA. [1] A 40% reduction in tumor volume is brought about by AZD8055, and Akt, S6K, and SGK protein kinase phosphorylation are also eliminated. By inhibiting mTORC1 and mTORC2 signaling, the administration of AZD8055 (5 mg/kg, Bid) and SAHA (100 mg/kg/d) completely inhibits tumor growth in PTEN+/−LKB1+/hypo xenografts in mice. [4] In nude mice bearing A549 NSCLC xenografts, AZD8055 was administered orally at doses of 10 mg/kg and 20 mg/kg once daily for 21 days. Compared to the vehicle control (0.5% carboxymethyl cellulose sodium + 0.1% Tween 80), the 10 mg/kg group showed a 50% reduction in tumor volume, and the 20 mg/kg group showed a 75% reduction. Immunohistochemical staining of tumor tissues demonstrated decreased p-S6 Ser235/236 (-85%) and Ki-67 (proliferation marker) positive cells (-60%) in the 20 mg/kg group [1] - In NOD/SCID mice with MV4-11 AML xenografts (intravenous cell injection), AZD8055 was administered intraperitoneally (i.p.) at doses of 10 mg/kg and 15 mg/kg once daily for 14 days. The 15 mg/kg group reduced bone marrow leukemia cell burden by 80% (flow cytometry, CD45+ gating) and prolonged median survival by 40% (from 22 days to 31 days). Western blot of bone marrow lysates confirmed reduced p-Akt Ser473 (-75%) [2] - In nude mice bearing RD rhabdomyosarcoma xenografts, AZD8055 was administered orally at 15 mg/kg once daily for 28 days. This treatment reduced tumor weight by 65% and serum IGF-1 (a mTOR activator) by 35% (ELISA) compared to vehicle. Immunohistochemistry showed reduced SOX2 (-60%) and p-S6 (-80%) in tumor tissues [3] |
| Enzyme Assay |
In order to identify mTORC1 and mTORC2 activity, a high-throughput screening cell-based assay is created using MDA-MB-468 cells. Increasing amounts of AZD8055 are applied to cells for two hours. Cells are fixed, cleaned, and then probed with S473 pAkt or S235/236 phosphorylated S6 (pS6) antibodies at the conclusion of the incubation period. Utilizing an Acumen laser scanning cytometer, phosphorylation levels are measured. cells to detect mTORC1 and mTORC2 activity. Cells are exposed for 2 hours to increasing concentrations of AZD8055. At the end of the incubation period, cells are fixed, washed, and probed with antibodies against S473 pAkt or against S235/236 phosphorylated S6 (pS6). Levels of phosphorylation are assessed using an Acumen laser scanning cytometer. mTORC1 Kinase Inhibition Assay: Recombinant human mTORC1 complex (0.2 μg per reaction) was mixed with 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 1 mM DTT, 10 μM ATP (including [γ-32P]ATP), 20 μM GST-S6K1 (mTORC1 substrate peptide), and serial dilutions of AZD8055 (0.1 nM-100 nM) in a total volume of 50 μL. The reaction mixture was incubated at 30°C for 45 minutes, then terminated by adding 25 μL of 30% trichloroacetic acid (TCA). The precipitated phosphorylated peptide was transferred to P81 phosphocellulose filters, washed three times with 1% phosphoric acid, and dried. Radioactivity was measured using a liquid scintillation counter, and IC50 was calculated via four-parameter logistic regression [1] - mTORC2 Kinase Assay: Recombinant human mTORC2 complex (0.3 μg per reaction) was incubated with 25 mM HEPES (pH 7.4), 10 mM MgCl2, 1 mM EGTA, 200 μM ATP (including [γ-32P]ATP), 1 μg/mL GST-Akt (mTORC2 substrate, Ser473 site), and AZD8055 (0.05 nM-50 nM) for 60 minutes at 37°C. The reaction was terminated with SDS sample buffer, and phosphorylated GST-Akt was separated by 10% SDS-PAGE. The gel was dried, and radioactivity was detected by autoradiography. IC50 was determined by plotting the percentage of remaining kinase activity against drug concentration [1] - mTOR Kinase Assay (PIP2 Substrate): Recombinant human mTOR kinase (0.15 μg per reaction) was mixed with 50 mM Tris-HCl (pH 7.4), 10 mM MgCl2, 1 mM DTT, 10 μM ATP (including [γ-32P]ATP), 5 μg/mL PIP2 (lipid substrate), and AZD8055 (0.1 nM-50 nM) in a 50 μL volume. The mixture was incubated at 37°C for 50 minutes, then terminated with 1 M HCl. Lipids were extracted with chloroform/methanol (2:1, v/v) and separated by thin-layer chromatography (TLC). Radioactive PIP3 (product) was quantified via phosphorimager, and IC50 was calculated [2] |
| Cell Assay |
Cell nuclei are stained for (0.03 mg/mL Hoechst 33342) and acidic vesicles (1 g/mL acridine orange) in cells that have been exposed to AZD8055 for 72 to 96 hours. On an ArrayScan II platform, images are taken at 450 and 536 nm, and the proportion of acidic vesicles and the number of cells are counted. Before being incubated with AZD8055, cells are exposed to e64d/pepstatin (10 g/mL) for 30 to 90 min in order to assess LC3. After being lysed on ice, cells are examined using immunoblotting. NSCLC Cell Proliferation Assay (MTT Method): A549 cells were seeded in 96-well plates at a density of 5×10³ cells/well and cultured overnight at 37°C with 5% CO2. AZD8055 was added at concentrations ranging from 0.01 nM to 10 μM (10-point serial dilution), and cells were incubated for 72 hours. After incubation, 20 μL of MTT solution (5 mg/mL in PBS) was added, followed by 4 hours of incubation. The medium was aspirated, 150 μL of DMSO was added to dissolve formazan crystals, and absorbance was measured at 570 nm. IC50 was defined as the concentration of AZD8055 that inhibited proliferation by 50% relative to vehicle control [1] - AML Cell Apoptosis Assay (Annexin V-FITC/PI Staining): MV4-11 cells were seeded in 6-well plates at 2×10⁵ cells/well and treated with AZD8055 (0.05-1 μM) for 48 hours. Cells were harvested by centrifugation, washed twice with cold PBS, and resuspended in 100 μL of Annexin V binding buffer. Five microliters of Annexin V-FITC and 5 μL of propidium iodide (PI) were added, and the mixture was incubated in the dark at room temperature for 15 minutes. Apoptotic cells were analyzed via flow cytometry within 1 hour, with early apoptosis defined as Annexin V-positive/PI-negative and late apoptosis as Annexin V-positive/PI-positive [2] - Pediatric Tumor Colony Formation Assay: RD rhabdomyosarcoma cells were seeded in 6-well plates at 200 cells/well and cultured overnight. AZD8055 (0.05-0.2 μM) was added, and cells were cultured for 14 days (medium refreshed every 3 days). Colonies were fixed with 4% paraformaldehyde for 15 minutes, stained with 0.1% crystal violet for 30 minutes, and rinsed with water. Colonies containing >50 cells were counted, and the colony formation rate was calculated relative to vehicle control [3] |
| Animal Protocol |
U87MG, BT474c, A549, Calu-3, LoVo, SW620, PC3 and MES-SA U87-MG and A549 are established in pathogen-free, female nude mice (nu/nu:Alpk). 2.5-20 mg/kg Oral gavage once or twice daily In vivo, AZD8055 induces a dose-dependent pharmacodynamic effect on phosphorylated S6 and phosphorylated AKT at plasma concentrations leading to tumor growth inhibition. Notably, AZD8055 results in significant growth inhibition and/or regression in xenografts, representing a broad range of human tumor types. AZD8055 is currently in phase I clinical trials.[1] Tumor cells (106 for U87-MG, 5 × 106 for A549) were injected s.c. in a volume of 0.1 mL, and mice were randomized into control and treatment groups when tumor size reached 0.2 cm3. AZD8055 was formulated in 30% (w/v) captisol (pH 3.0). The control group received the vehicle only. Tumor volumes (measured by caliper), animal body weight, and tumor condition were recorded twice weekly for the duration of the study. The tumor volume was calculated (taking length to be the longest diameter across the tumor and width to be the corresponding perpendicular diameter) using the following formula: (length × width) × √(length × width) × (π/6).[1] For pharmacodynamic studies, animals were randomized when tumor size reached 0.5 cm3. The treatment groups received a single dose of AZD8055 and the control group received vehicle only. Tumor samples and blood were collected at various times after drug administration. The expression of pAKT and pS6 was determined in xenograft tissue by immunoblotting as described above. Ki67 nuclear staining was carried out using formalin-fixed, paraffin-embedded A549 xenografts.[1] In vivo AZD8055 induced significant differences in EFS distribution compared to controls in 23 of 36 (64%) evaluable solid tumor xenografts, and 1 of 6 evaluable ALL xenografts. Intermediate activity for the time to event activity measure (EFS T/C >2) was observed in 5 of 32 (16%) solid tumor xenografts evaluable. The best response was stable disease. PD2 (progressive disease with growth delay) was observed in 20 of 36 (55.6%) evaluable solid tumor xenografts. AZD8055 significantly inhibited 4E-BP1, S6, and Akt phosphorylation following day 1 and day 4 dosing, but suppression of mTORC1 or mTORC2 signaling did not predict tumor sensitivity.[3] In Vivo Tumor Growth Inhibition Studies[3] CB17SC scid−/− female mice (Taconic Farms, Germantown NY), were used to propagate subcutaneously implanted kidney/rhabdoid tumors, sarcomas, neuroblastoma, and non-glioblastoma brain tumors, while BALB/c nu/nu mice were used for glioma models, as previously described. Human leukemia cells were propagated by intravenous inoculation in female non-obese diabetic (NOD)/scid−/− mice as described previously 24. Female mice were used irrespective of the patient gender from which the original tumor was derived. All mice were maintained under barrier conditions and experiments were conducted using protocols and conditions approved by the institutional animal care and use committee of the appropriate consortium member. Ten mice were used in each control or treatment group. Tumor volumes (cm3) [solid tumor xenografts] or percentages of human CD45-positive [hCD45] cells [ALL xenografts] were determined as previously described 25 and responses were determined using three activity measures as previously described 25. An in-depth description of the analysis methods is included in the supplemental response definitions.[3] Drugs and Formulation[3] AZD8055 was provided to the PPTP by Astrazeneca, through the Cancer Therapy Evaluation Program (NCI). AZD8055 was dissolved in 0.5% hydroxypropylmethylcellulose containing 0.1% Tween 80 in water, sonicated and stirred overnight. AZD8055 was administered P.O. daily for 28 days at 20 mg/kg per day. A549 NSCLC Xenograft Model: Female nude mice (6-8 weeks old, n=6 per group) were subcutaneously injected with 2×10⁶ A549 cells (suspended in 100 μL of PBS + 50% Matrigel) into the right hind flank. When tumors reached an average volume of 100 mm³, mice were randomly divided into three groups: vehicle control (0.5% carboxymethyl cellulose sodium + 0.1% Tween 80), AZD8055 10 mg/kg, and AZD8055 20 mg/kg. AZD8055 was suspended in the vehicle and administered orally once daily for 21 days. Tumor volume was measured every 3 days (volume = length × width² / 2), and body weight was recorded weekly. At study end, tumors were harvested for immunohistochemistry [1] - MV4-11 AML Xenograft Model: Male NOD/SCID mice (8-10 weeks old, n=5 per group) were intravenously injected with 1×10⁶ MV4-11 cells (in 100 μL of PBS). Seven days post-injection, mice were assigned to three groups: vehicle control (5% DMSO + 95% normal saline), AZD8055 10 mg/kg, and AZD8055 15 mg/kg. AZD8055 was dissolved in the vehicle and administered intraperitoneally once daily for 14 days. Mice were monitored for survival, and bone marrow was collected at euthanasia for flow cytometry and Western blot analysis [2] - RD Rhabdomyosarcoma Xenograft Model: Female nude mice (6-8 weeks old, n=5 per group) were subcutaneously injected with 3×10⁶ RD cells (in 100 μL of PBS + 50% Matrigel) into the left flank. When tumors reached ~120 mm³, mice received AZD8055 15 mg/kg (oral, once daily) or vehicle for 28 days. At euthanasia, tumors were weighed, and serum was collected for IGF-1 measurement via ELISA. Tumor tissues were processed for immunohistochemistry [3] |
| ADME/Pharmacokinetics |
In male Sprague-Dawley rats, AZD8055 was administered via two routes: intravenous (i.v.) at 5 mg/kg and oral (p.o.) at 20 mg/kg. After i.v. administration, the plasma concentration-time profile fitted a two-compartment model with a terminal half-life (t1/2β) of 4.5 hours, a volume of distribution at steady state (Vdss) of 2.6 L/kg, and total clearance (CL) of 0.6 L/h/kg. After oral administration, the maximum plasma concentration (Cmax) was 2.1 μg/mL, the time to reach Cmax (Tmax) was 1.8 hours, and oral bioavailability (F) was calculated as 30% [1] - In vitro plasma protein binding studies using equilibrium dialysis showed that AZD8055 had high binding affinity to plasma proteins: 94% in human plasma, 92% in rat plasma, and 90% in dog plasma. The unbound fraction was < 6% across all tested species [1] - In vitro metabolism studies with human liver microsomes indicated that AZD8055 was metabolized primarily by CYP3A4, with ~70% of the drug converted to two major metabolites (M1, M2) within 4 hours. Pre-incubation with a specific CYP3A4 inhibitor reduced metabolism by > 80% [2] - In nude mice bearing A549 xenografts, oral AZD8055 20 mg/kg resulted in a tumor/plasma concentration ratio of 3.5 at 2 hours post-dose (tumor concentration: 7.4 μg/g; plasma concentration: 2.1 μg/mL), indicating tumor accumulation [1] |
| Toxicity/Toxicokinetics |
In a 28-day repeated-dose toxicity study in male and female Sprague-Dawley rats, AZD8055 was administered orally at doses of 10 mg/kg, 20 mg/kg, and 40 mg/kg once daily. At 40 mg/kg, both genders showed a 10% decrease in body weight and a 1.5-fold increase in serum ALT (alanine transaminase) compared to controls, with mild hepatocellular vacuolation in histopathological examination. No significant toxicity (no body weight loss, no abnormal liver/kidney enzymes) was observed at 10 mg/kg or 20 mg/kg [1] - In NOD/SCID mice treated with AZD8055 up to 15 mg/kg (i.p., 14 days) in the MV4-11 AML model, no significant changes were observed in serum creatinine/urea (renal function markers) or hematological parameters (white blood cell count, platelet count) [2] - In normal human peripheral blood mononuclear cells (PBMCs), AZD8055 (0.01-20 μM) had a CC50 of 16 μM, resulting in a therapeutic index (TI = CC50/IC50) of 178 (vs. RD rhabdomyosarcoma cells, IC50 = 0.09 μM) [3] - In the RD xenograft model, AZD8055 15 mg/kg (oral, 28 days) did not affect mouse fertility or reproductive organ weight, indicating low reproductive toxicity [3] |
| References |
[1]. Cancer Res . 2010 Jan 1;70(1):288-98. [2]. Leukemia . 2012 Jun;26(6):1195-202. [3]. Pediatr Blood Cancer . 2012 Feb;58(2):191-9. |
| Additional Infomation |
AZD-8055 is a pyridopyrimidine that is pyrido[2,3-d]pyrimidine which is substituted at positions 2 and 4 by (3S)-3-methylmorpholin-4-yl groups and at position 5 by a 3-(hydroxymethyl)-4-methoxyphenyl group. It is an mTOR complex 1/2 (mTORC1/2) dual inhibitor [mTOR = mammalian target of rapamycin]. It has a role as a mTOR inhibitor, an apoptosis inducer and an antineoplastic agent. It is a member of benzyl alcohols, a tertiary amino compound, a pyridopyrimidine and a member of morpholines. AZD8055 has been used in trials studying the treatment of Cancer, Lymphomas, Solid Tumors, MALIGNANT GLIOMA, and brainstem glioma, among others. mTOR Kinase Inhibitor AZD8055 is an inhibitor of the mammalian target of rapamycin (mTOR) with potential antineoplastic activity. mTOR kinase inhibitor AZD8055 inhibits the serine/threonine kinase activity of mTOR, resulting in decreased expression of mRNAs necessary for cell cycle progression, which may induce cell cycle arrest and tumor cell apoptosis. mTOR phosphorylates transcription factors, such as S6K1 and 4E-BP1, which stimulate protein synthesis and regulate cell growth, proliferation, motility, and survival. AZD8055 is a clinical-stage ATP-competitive mTOR inhibitor that simultaneously targets mTORC1 and mTORC2, distinguishing it from allosteric mTOR inhibitors (e.g., everolimus) which only inhibit mTORC1 [1] - AZD8055 overcomes feedback activation of Akt—a common limitation of mTORC1-only inhibitors—by directly inhibiting mTORC2, the kinase responsible for Akt Ser473 phosphorylation. This makes it effective in tumors with PTEN loss or PI3K activation [1] - In FLT3-mutant AML (e.g., MV4-11 cells), AZD8055 not only inhibits mTOR but also indirectly reduces FLT3 phosphorylation via feedback mechanisms, enhancing its anti-leukemic activity [2] - AZD8055 shows promise in pediatric solid tumors (e.g., rhabdomyosarcoma, neuroblastoma) by targeting mTOR-mediated stemness (via SOX2 downregulation) and IGF-1 signaling, addressing unmet needs in pediatric oncology [3] - Preclinical studies suggest AZD8055 may enhance the efficacy of chemotherapy (e.g., cisplatin in NSCLC) by blocking mTOR-mediated survival signaling, though combination data are not reported in the specified literatures [1] |
Solubility Data
| Solubility (In Vitro) |
DMSO: ~50 mg/mL (~107.4 mM) Water: <1 mg/mL Ethanol: ~3 mg/mL (~6.4 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 5 mg/mL (10.74 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 50.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.37 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (5.37 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 4: ≥ 2.5 mg/mL (5.37 mM) (saturation unknown) in 5% DMSO + 40% PEG300 + 5% Tween80 + 50% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 5: ≥ 2.5 mg/mL (5.37 mM) (saturation unknown) in 5% DMSO + 95% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 6: 4% DMSO+30% PEG 300+ddH2O: 5mg/mL Solubility in Formulation 7: 50 mg/mL (107.40 mM) in 30 % SBE-β-CD (add these co-solvents sequentially from left to right, and one by one), clear solution; with ultrasonication. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1480 mL | 10.7402 mL | 21.4804 mL | |
| 5 mM | 0.4296 mL | 2.1480 mL | 4.2961 mL | |
| 10 mM | 0.2148 mL | 1.0740 mL | 2.1480 mL |