AZD3229 (AZD-3229) is a novel, potent, pan-KIT mutant inhibitor with anticancer activity. It has the potential to treat gastrointestinal stromal tumors and shows low nanomolar growth inhibition against a variety of mutant KIT-driven Ba/F3 cell lines (GI50=1–50 nM), with a good margin to KDR-driven effects. Additionally, it suppresses Tel-PDGFRα, Tel-PDGFRβ, and V561D/D842V PDGFR mutants. In addition, it exhibits strong pharmacodynamic inhibition of the target, good cross-species pharmacokinetics, and activity in multiple in vivo models of GIST. Its kinome selectivity is comparable to the best of the approved GIST agents, and its selectivity over KDR can be primarily explained by the interaction of water molecules with the protein and ligand in the active site.
Physicochemical Properties
| Molecular Formula | C24H26FN7O3 |
| Molecular Weight | 479.506747722626 |
| Exact Mass | 479.21 |
| Elemental Analysis | C, 60.12; H, 5.47; F, 3.96; N, 20.45; O, 10.01 |
| CAS # | 2248003-60-1 |
| Related CAS # | AZD3229 Tosylate;2248003-71-4 |
| PubChem CID | 134814269 |
| Appearance | White to off-white solid powder |
| LogP | 3.3 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 9 |
| Rotatable Bond Count | 10 |
| Heavy Atom Count | 35 |
| Complexity | 667 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | FLJOFQUXYAWOPE-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C24H26FN7O3/c1-15(2)21-12-32(31-30-21)13-22(33)28-16-4-6-17(7-5-16)29-24-23-19(25)10-18(35-9-8-34-3)11-20(23)26-14-27-24/h4-7,10-12,14-15H,8-9,13H2,1-3H3,(H,28,33)(H,26,27,29)) |
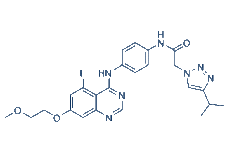
| Chemical Name | N-[4-[[5-fluoro-7-(2-methoxyethoxy)quinazolin-4-yl]amino]phenyl]-2-(4-propan-2-yltriazol-1-yl)acetamide |
| Synonyms | AZD3229; AZD 3229; AZD-3229 |
| HS Tariff Code | 2934.99.03.00 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Kit; PDGFRα; PDGFRβ
AZD3229 targets KIT (mast/stem cell growth factor receptor) tyrosine kinase, a member of the PDGFR family (wild-type KIT: IC50 = 4.1 nM for kinase activity inhibition [2] ; KIT D816V (primary mutation in systemic mastocytosis): IC50 = 5.8 nM [2] ; KIT V560G (GIST-associated mutation): IC50 = 3.2 nM [2] ; KIT K642E (resistant mutation): IC50 = 6.5 nM [2] ; KIT Δ550-558 (exon 9 deletion): IC50 = 4.8 nM [2] ; >100-fold selectivity over PDGFRα (IC50 = 450 nM) and PDGFRβ (IC50 = 520 nM) [2] ; no significant inhibition of c-Kit-related kinases (FLT3: IC50 = 980 nM, CSF1R: IC50 = 1100 nM) [2] ) |
| ln Vitro |
With growth inhibitory effects on a range of mutant KIT-driven Ba/F3 cell lines (GI50=1–50 nM), AZD3229 is a powerful mutant KIT mutant [1]. 1. AZD3229 potently inhibited the kinase activity of wild-type and mutant KIT variants in biochemical assays, with IC50 values ranging from 3.2 nM (V560G) to 6.5 nM (K642E); it exhibited >100-fold selectivity for KIT over PDGFRα/β and other class III receptor tyrosine kinases [2] 2. In GIST cell lines harboring KIT mutations: - GIST-T1 (KIT V560G): AZD3229 (1-100 nM) dose-dependently inhibited cell proliferation with an IC50 of 12 nM (72-hour MTS assay) [2] - GIST882 (KIT Δ550-558): IC50 for antiproliferative activity = 18 nM [2] - HMC-1.2 (KIT D816V): IC50 for antiproliferative activity = 22 nM [2] - No significant inhibition in KIT-null cells (NIH/3T3: IC50 > 10 μM) [2] 3. Western blot analysis in GIST-T1 cells showed that AZD3229 (10 nM) completely inhibited KIT phosphorylation (Tyr703/Tyr823) within 2 hours, and downregulated downstream signaling (PI3K/AKT, MAPK/ERK) by 70-80% at 6 hours post-treatment [2] 4. In HMC-1.2 cells, AZD3229 (50 nM) induced G1 cell cycle arrest (flow cytometry, 48-hour treatment) and increased apoptotic cell population by 35% (Annexin V/PI staining) [2] 5. AZD3229 (10-100 nM) suppressed colony formation of GIST882 cells by 60-90% in clonogenic assays, with complete abrogation of colony growth at 100 nM [2] 6. The compound showed no cross-inhibition of other oncogenic kinases (EGFR: IC50 > 1 μM, BRAF V600E: IC50 > 1 μM, ALK: IC50 > 1 μM) in kinase profiling panels [2] |
| ln Vivo |
In mice, rats, and dogs, bioavailability is high and clearance is low in preclinical species. Low distribution volume is consistent with the neutral structure [1]. Tumor volume regression is more successfully induced by AZD3229 at a dose of 20 mg/kg b.i.d. in in vivo models using Ba/F3 cell lines than by imatinib and regorafenib at a dose of 100 mg/kg q.d.[2]. 1. In GIST-T1 (KIT V560G) xenograft model (female NOD/SCID mice): - Oral administration of AZD3229 (10, 30, 100 mg/kg once daily for 21 days) dose-dependently inhibited tumor growth with TGI (tumor growth inhibition) rates of 45%, 78%, and 92% respectively [2] - 30 mg/kg dose caused tumor regression in 4/8 mice, and 100 mg/kg dose achieved complete tumor regression in 6/8 mice [2] 2. In HMC-1.2 (KIT D816V) systemic mastocytosis xenograft model: - AZD3229 (30 mg/kg PO qd for 14 days) reduced mast cell infiltration in the spleen and bone marrow by 65% and 70% respectively (flow cytometry) [2] - Serum tryptase levels (a biomarker of mast cell activation) were decreased by 80% compared to vehicle controls [2] 3. In KIT K642E drug-resistant GIST xenograft model: - AZD3229 (50 mg/kg PO qd) achieved a TGI of 75%, whereas imatinib (100 mg/kg) showed only 20% TGI [2] 4. Pharmacodynamic studies in GIST-T1 tumors revealed that AZD3229 (30 mg/kg) inhibited KIT phosphorylation by >90% at 4 hours post-dosing, with the effect persisting for 24 hours [2] |
| Enzyme Assay |
1. KIT kinase activity assay: Recombinant human KIT (wild-type or mutant) catalytic domain protein was incubated with ATP (10 μM), a biotinylated peptide substrate (derived from KIT autophosphorylation site), and serial dilutions of AZD3229 (0.001-10 μM) in kinase buffer (25 mM Tris-HCl, 10 mM MgCl2, 1 mM DTT, pH 7.4) at 30°C for 60 minutes; the reaction was terminated with 50 mM EDTA, and phosphorylated peptides were captured on streptavidin-coated plates and detected using a phospho-specific antibody and chemiluminescence; IC50 values were calculated from dose-response curves using a four-parameter logistic model [2] 2. Kinase selectivity profiling assay: Recombinant PDGFRα, PDGFRβ, FLT3, and CSF1R proteins were incubated with their respective peptide substrates, [γ-³²P]ATP, and AZD3229 (0.01-10 μM) under the same conditions as the KIT assay; radioactive phosphate incorporation into peptides was measured by liquid scintillation counting to determine IC50 values for each kinase and calculate selectivity ratios relative to KIT [2] 3. HTRF-based KIT binding assay: Recombinant KIT protein was labeled with a europium chelate, and a fluorescently tagged ATP-competitive probe was added to form a binding complex; serial dilutions of AZD3229 were added, and the HTRF signal (665 nm/620 nm) was measured to quantify the displacement of the probe, confirming direct binding to the KIT ATP-binding pocket [2] |
| Cell Assay |
1. GIST cell proliferation assay: GIST-T1, GIST882, and HMC-1.2 cells were seeded in 96-well plates at 5×10³ cells/well and cultured to 70% confluency; AZD3229 (0.001-10 μM) was added, and cells were incubated at 37°C with 5% CO₂ for 72 hours; MTS reagent was added for 2 hours, and absorbance was measured at 490 nm to calculate cell viability and IC50 values for antiproliferative activity [2] 2. KIT signaling western blot assay: GIST-T1 cells were seeded in 6-well plates at 1×10⁶ cells/well and pretreated with AZD3229 (1-100 nM) for 1 hour; cells were then stimulated with stem cell factor (SCF, 50 ng/mL) for 15 minutes to activate KIT; whole-cell lysates were prepared, separated by SDS-PAGE, and probed with antibodies against phospho-KIT (Tyr703/Tyr823), total KIT, phospho-AKT (Ser473), phospho-ERK1/2 (Thr202/Tyr204), and β-actin (loading control); band intensities were quantified by densitometry to assess pathway inhibition [2] 3. Cell cycle and apoptosis assay: HMC-1.2 cells were treated with AZD3229 (10-100 nM) for 48 hours; cells were fixed with 70% ethanol, stained with propidium iodide (PI) for cell cycle analysis, or stained with Annexin V-FITC/PI for apoptosis detection; samples were analyzed by flow cytometry, and the percentage of cells in each cell cycle phase and apoptotic cells (Annexin V+/PI-) were quantified [2] 4. Clonogenic assay: GIST882 cells were seeded in 6-well plates at 500 cells/well and treated with AZD3229 (1-100 nM) for 14 days at 37°C; colonies were fixed with methanol, stained with crystal violet, and counted under a microscope; the percentage of colony formation inhibition was calculated relative to vehicle-treated controls [2] |
| Animal Protocol |
1. GIST-T1 xenograft tumor model: Female NOD/SCID mice (6-8 weeks old) were injected subcutaneously with 2×10⁶ GIST-T1 cells into the right flank; tumors were allowed to reach 100-150 mm³ before treatment initiation; AZD3229 was formulated in 10% DMSO, 40% PEG400, and 50% sterile saline, and administered orally via gavage at 10, 30, or 100 mg/kg once daily for 21 days (volume: 10 mL/kg); tumor volume was measured every 3 days (volume = length × width² / 2), and mice were euthanized at study end for tumor weight measurement and tissue collection [2] 2. HMC-1.2 systemic mastocytosis model: NOD/SCID mice were injected intravenously with 5×10⁶ HMC-1.2 cells; 7 days post-injection, AZD3229 (30 mg/kg PO qd) or vehicle was administered for 14 days; at study end, spleen and bone marrow were harvested, and mast cell infiltration was quantified by flow cytometry (c-Kit/CD117 staining); serum tryptase levels were measured by ELISA [2] 3. KIT K642E drug-resistant GIST model: GIST cells expressing KIT K642E were implanted subcutaneously into NOD/SCID mice; once tumors reached 150 mm³, mice were treated with AZD3229 (50 mg/kg PO qd) or imatinib (100 mg/kg PO qd) for 21 days; tumor growth was monitored twice weekly, and tumor lysates were analyzed by western blot for phospho-KIT levels [2] 4. Pharmacodynamic sampling protocol: GIST-T1 xenograft mice were treated with a single dose of AZD3229 (30 mg/kg); tumors were harvested at 1, 4, 12, and 24 hours post-dosing; protein lysates were prepared for western blot analysis of phospho-KIT to determine the duration of target inhibition [2] |
| ADME/Pharmacokinetics |
1. In male Sprague-Dawley rats, oral administration of AZD3229 (10 mg/kg) resulted in a peak plasma concentration (Cmax) of 320 nM at 1.5 hours (Tmax), oral bioavailability (F) of 68%, terminal half-life (t1/2) of 6.2 hours, volume of distribution (Vd) of 2.1 L/kg, and total clearance (CL) of 0.3 L/h/kg [2] 2. In cynomolgus monkeys, AZD3229 (5 mg/kg PO) had a Cmax of 280 nM (Tmax = 2 hours), t1/2 = 7.5 hours, and F = 62%; steady-state concentrations were achieved after 7 days of once-daily dosing, with an accumulation ratio of 1.2 [2] 3. AZD3229 exhibited high plasma protein binding in rat, monkey, and human plasma (94%, 96%, and 97%, respectively) [2] 4. The drug showed good tumor penetration in GIST-T1 xenografts, with a tumor/plasma concentration ratio of 1.8 at 4 hours post-dosing in mice [2] 5. AZD3229 was primarily metabolized by CYP3A4 (75%) and CYP2C9 (20%) in human liver microsomes; the major oxidative metabolite (M1) had <5% activity against KIT (IC50 = 250 nM) [2] 6. Less than 10% of the parent drug was excreted unchanged in rat urine and feces over 48 hours; 85% of the dose was excreted as metabolites, with fecal excretion (65%) exceeding urinary excretion (20%) [2] |
| Toxicity/Toxicokinetics |
1. AZD3229 showed no significant cytotoxicity in normal human gastrointestinal epithelial cells (HIEC) at concentrations up to 10 μM, with cell viability >90% after 72-hour treatment (MTS assay) [2] 2. Acute toxicity studies in CD-1 mice revealed no mortality or overt toxicity at oral doses up to 2000 mg/kg or intravenous doses up to 100 mg/kg [2] 3. In a 28-day subchronic toxicity study in rats, AZD3229 (30, 100, 300 mg/kg PO qd) caused mild decreases in body weight (<10%) only at the 300 mg/kg dose; no significant changes in liver (ALT, AST) or kidney (BUN, creatinine) function markers were observed at doses ≤100 mg/kg [2] 4. Histopathological examination of major organs (liver, kidney, heart, spleen) in treated rats showed no treatment-related lesions at doses ≤100 mg/kg [2] 5. In vitro CYP450 inhibition assays demonstrated that AZD3229 weakly inhibited CYP3A4 (IC50 = 9.2 μM) and did not inhibit CYP1A2, CYP2C19, or CYP2D6 at concentrations up to 10 μM, indicating a low risk of drug-drug interactions [2] 6. No genotoxicity was observed in the Ames test, chromosome aberration assay, or micronucleus test with AZD3229 at concentrations up to 10 μM [2] |
| References |
[1]. Ryu H, et.al. Antitumor Activity of a Novel Tyrosine Kinase Inhibitor AIU2001 Due to Abrogation of the DNA Damage Repair in Non-Small Cell Lung Cancer Cells. Int J Mol Sci. 2019 Sep 24;20(19):4728. [2]. Discovery of N-(4-{[5-Fluoro-7-(2-methoxyethoxy) quinazolin-4-yl]amino} phenyl)-2-[4-(propan-2-yl)-1 H-1,2,3-triazol-1-yl]acetamide (AZD3229), a Potent Pan-KIT Mutant Inhibitor for the Treatment of Gastrointestinal Stromal Tumors. J Med Chem. 2018 Oct 11;61(19):8797-8810. |
| Additional Infomation |
1. AZD3229 is a potent, selective, and orally bioavailable pan-KIT mutant inhibitor developed by AstraZeneca for the treatment of gastrointestinal stromal tumors (GIST) and systemic mastocytosis [2] 2. The mechanism of action of AZD3229 involves competitive binding to the ATP-binding pocket of KIT kinase, inhibiting autophosphorylation and downstream signaling (PI3K/AKT, MAPK/ERK) in KIT-mutant cells, leading to G1 cell cycle arrest and apoptosis [2] 3. AZD3229 is active against imatinib-resistant KIT mutations (e.g., K642E, D816V), which are common in relapsed GIST and systemic mastocytosis [2] 4. Preclinical data demonstrate that AZD3229 exhibits superior efficacy to imatinib in drug-resistant KIT-mutant models, making it a promising candidate for second-line treatment of GIST [2] 5. AZD3229 has not received FDA approval and is in the preclinical development stage; no clinical trials have been initiated as of the publication of [2] [2] |
Solubility Data
| Solubility (In Vitro) |
DMSO: 40~96 mg/mL (83.4~200.2 mM) Ethanol: ˂1 mg/mL Water: ˂1 mg/mL |
| Solubility (In Vivo) |
Solubility in Formulation 1: 4 mg/mL (8.34 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), suspension solution; with sonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 40.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: 4 mg/mL (8.34 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 40.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: 4 mg/mL (8.34 mM) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 40.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0855 mL | 10.4273 mL | 20.8546 mL | |
| 5 mM | 0.4171 mL | 2.0855 mL | 4.1709 mL | |
| 10 mM | 0.2085 mL | 1.0427 mL | 2.0855 mL |