Physicochemical Properties
| Molecular Formula | C18H21N5O2S |
| Molecular Weight | 371.4566 |
| Exact Mass | 371.141 |
| CAS # | 602306-29-6 |
| Related CAS # | 602306-29-6 |
| PubChem CID | 16747683 |
| Appearance | White to light yellow solid powder |
| Density | 1.3±0.1 g/cm3 |
| Boiling Point | 655.2±65.0 °C at 760 mmHg |
| Flash Point | 350.1±34.3 °C |
| Vapour Pressure | 0.0±2.0 mmHg at 25°C |
| Index of Refraction | 1.648 |
| LogP | 2.13 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 6 |
| Rotatable Bond Count | 5 |
| Heavy Atom Count | 26 |
| Complexity | 556 |
| Defined Atom Stereocenter Count | 0 |
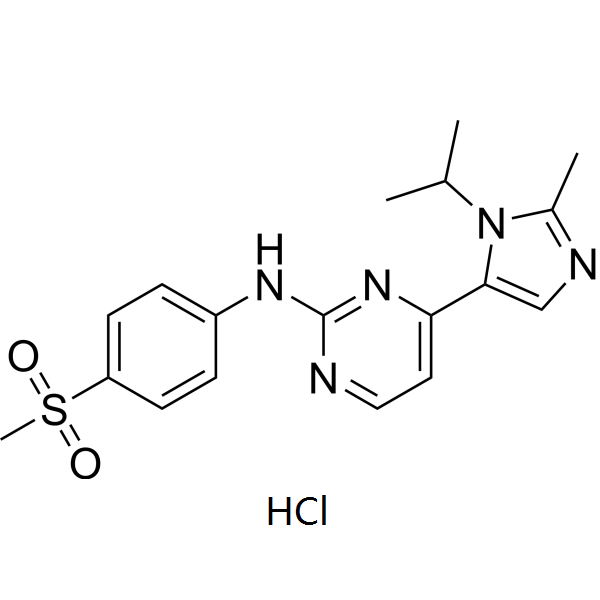
| SMILES | O=S(C1=CC=C(NC2=NC=CC(C3=CN=C(C)N3C(C)C)=N2)C=C1)(C)=O |
| Synonyms | AZD5438 HCl; AZD-5438 HCl; 602306-29-6; AZD-5438; DTXSID501025680; 2-PYRIMIDINAMINE, 4-(2-METHYL-1-(1-METHYLETHYL)-1H-IMIDAZOL-5-YL)-N-(4-(METHYLSULFONYL)PHENYL)-; 2-Pyrimidinamine, 4-[2-methyl-1-(1-methylethyl)-1H-imidazol-5-yl]-N-[4-(methylsulfonyl)phenyl]-; AZD 5438 HCl |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | cdk2-cyclin E (IC50 = 6 nM); cdk2-cyclin A (IC50 = 45 nM); cdk5-p25 (IC50 = 14 nM); cdk1-cyclin B1 (IC50 = 16 nM); cdk9-cyclin T (IC50 = 20 nM); cdk6-cyclin D3 (IC50 = 21 nM); cdk4-cyclin D1 (IC50 = 449 nM); cdk7-cyclin H (IC50 = 821 nM) |
| ln Vitro | Cyclin E-cdk2, Cyclin A-cdk2, Cyclin B1-cdk1, p25-cdk5, Cyclin D3-cdk6, and Cyclin T-cdk9 are all efficiently inhibited by AZD5438. activity (16, 21, 20 nM, 6, 45 nM). Cyclin E-cdk2, Cyclin A-cdk2, Cyclin B1-cdk1, p25-cdk5, Cyclin D3-cdk6, and Cyclin T-cdk9 are all potently inhibited by AZD5438. Similar to numerous other CDK regulators, AZD5438 suppresses p25-cdk5 and glycogen synthase, which in turn control the sporadic activity of 3β (IC50 values of 14 and 17 nM, correspondingly) [1]. The cellular radiation targeting of NSCLC cells is greatly improved by AZD5438. AZD5438 can also improve tumor growth delay when used in conjunction with radiation therapy, with an improved cell range of 1.2–1.7 [2]. |
| ln Vivo | AZD5438 (50 mg/kg twice day or 75 mg/kg for oncology) prevents human xenografts from growing. AZD5438 lowers the percentage of circulating active cells internally. Additional pharmacodynamic investigation of SW620 xenografts treated with AZD5438 revealed that after a single threshold of 16 hours, effective dosages of AZD5438 (>40% tumor growth suppression) maintained inhibitory effects on biomarkers including phospho-pRbSer249/Thr252[1]. |
| Enzyme Assay |
A scintillation proximity assay is used to test AZD5438's capacity to inhibit CDK activity. It involves using recombinant CDK-cyclin complexes of cyclin-Ecdk2, cdk2-cyclin A, cdk4-cyclin D, and recombinant retinoblastoma substrate (amino acids 792-928) or cdk1-cyclin B1 with a peptide substrate derived from the in vitro p34cdc2 phosphorylation site of histone H1 (biotin-X-Pro-Lys-Thr-Pro-Lys-Lys-Ala-Lys-Lys-Leu). Using peptide substrate (AKKPKTPKKAKKLOH), AZD5438's activity against recombinant cdk5/p25 is assessed in a scintillation proximity assay-based assay at 2 μM ATP. By using human purified glycogen synthase kinase 3βenzyme and eukaryotic initiation factor 2B substrate (at 1 μM ATP), the scintillation proximity assay is used to determine the inhibition of glycogen synthase kinase 3β activity. AZD5438 is subjected to a kinase selectivity screening service in which it is screened against cdk6-cyclin D3, cdk7-cyclin H/MAT1 (cdk activating kinase complex), and cdk9-cyclin T. Recombinant CDK1/cyclin B, CDK2/cyclin A/E, and CDK9/cyclin T kinase complexes were prepared. Gradient concentrations of AZD5438 were mixed with kinase complexes, ATP substrate, and specific peptides, and incubated at 37°C for 60 minutes; the phosphorylation level of the substrate was determined by radioactive phosphorylation assay, and the IC50 value of each kinase was calculated by curve fitting according to the inhibitory effect of different drug concentrations [1] Fluorescence resonance energy transfer (FRET) was used to verify CDK2 activity: AZD5438 was mixed with CDK2/cyclin A complex, fluorescently labeled substrate peptide, and ATP. After reacting at 30°C for 45 minutes, the fluorescence signal intensity was detected to calculate the kinase activity inhibition rate [2] |
| Cell Assay |
Solid tumor cell lines are used to test AZD5438. In summary, AZD5438 is added to cells at different concentrations and incubated for 48 hours. After the incubation period, 5-bromo-2′-deoxyuridine (BrdUrd) is pulsed into the cells, and the quantity of DNA synthesis is quantified. Specifically, cell death has no bearing on the IC50 for proliferation inhibition. The following protocol is used to seed multiple myeloma cell lines into 96-well plates: RPMI 1640 supplemented with 10% FCS and glutamine, followed by a 72-hour dose of AZD5438. AlamarBlue is used to measure cell growth, and GI50 values are computed using the pretreatment control values. Tumor cells were seeded in 96-well plates (5×10³ cells/well) and cultured for 24 hours, then gradient concentrations of AZD5438 (0.01-10 μM) were added and cultured for another 72 hours; the CellTiter-Glo luminescent method was used to detect cell viability, and the cell survival rate was calculated to fit the curve for IC50 value [1] A549 cells were pretreated with AZD5438 (100 nM) for 2 hours, then irradiated with gradient doses of X-rays (0-8 Gy). After 14 days of culture, the clonogenic assay was used to detect the cell survival fraction, and the sensitization enhancement ratio (SER) was calculated [2] After treating cells with AZD5438 (100 nM) for 48 hours, the cells were collected and fixed, stained with PI, and the cell cycle distribution was analyzed by flow cytometry; total cellular protein was extracted, and the expression of histone H1, phosphorylated histone H1, Rb, phosphorylated Rb and other proteins was detected by Western blot [1] After treating MCF-7 cells with the drug for 72 hours, the Annexin V-FITC/PI double staining method was used, incubated at room temperature for 15 minutes, and the proportion of apoptotic cells was detected by flow cytometry; caspase-3 activity was determined by a caspase-3 activity assay kit, and the expression of PARP, Mcl-1 and other related proteins was detected by Western blot [1] Tumor cells were seeded in 6-well plates (1×10³ cells/well) and cultured for 24 hours, then AZD5438 (0.01-1 μM) was added and cultured for another 14 days; the medium was discarded, fixed with methanol, stained with crystal violet, and clones with more than 50 cells were counted to calculate the clonogenic rate [1] |
| Animal Protocol |
Every human tumor xenograft, with the exception of HX147, is created by subcutaneously injecting 100 μL of tumor cells (a mixture of 1×10 6 and 1×10 7 cells mixed 1:1 with Matrigel). The origin of HX147 tumors is fragment implants (1 mm 3 pieces) from tumors removed from mice that were first injected subcutaneously with 1×10 7 cells. Prior to being implanted for antitumor work, these tumor fragments undergo three passages through mice. As previously mentioned, tumor volumes are computed, tumor measurements are made up to three times a week using calipers, and the data are plotted as the geometric mean for each group versus time. Tumors that reach a mean size of roughly >0.2 cm 3 in mice and >0.5 cm 3 in rats are the triggers for randomization of animals into treatment groups (usually n=10). Hydroxy-propyl-methyl-cellulose is used to prepare AZD5438. Oral gavage of AZD5438 (37.5-75 mg/kg) or vehicle control is administered once or twice daily to animals for approximately three weeks in each scenario. As previously mentioned, the tumor volume and percentage tumor growth inhibition (% TGI) are computed. Any variation in tumor volume is statistically analyzed using the standard t test, with a significance level of P<0.05. Female nude mice (6-8 weeks old) were subcutaneously inoculated with A549 cell suspension (2×10⁶ cells/mouse) on the right back. Drug administration started when the tumor volume reached 100-150 mm³; AZD5438 was dissolved in normal saline containing 0.5% hydroxypropyl methylcellulose and 0.1% Tween 80, and administered orally at 50 mg/kg once daily for 21 days; tumor volume and mouse weight were measured every 3 days, and tumors were excised and weighed at the end of the experiment to detect CDK activity and related protein expression in tumor tissues [1] A549 nude mouse xenograft models (tumor volume reached 200 mm³) were divided into control group, radiotherapy alone group, AZD5438 monotherapy group, and combination group; the radiotherapy group received local tumor irradiation with 2 Gy X-rays twice a week for 3 weeks; AZD5438 was administered orally at 75 mg/kg once daily, starting synchronously with radiotherapy for 3 weeks; the tumor growth inhibition rate was calculated at the end of the experiment, and the expression of DNA damage marker (γ-H2AX) in tumor tissues was detected [2] Nude mice with MCF-7 xenograft models (tumor volume reached 120 mm³) were given oral AZD5438 at 60 mg/kg once daily for 14 days; mice were sacrificed 24 hours after the last administration, and tumor tissues were collected for protein extraction and Western blot analysis [1] |
| ADME/Pharmacokinetics | After oral administration of 50 mg/kg AZD5438 in rats, the time to peak concentration (Tmax)=1.8 hours, peak plasma concentration (Cmax)=980 ng/mL, and oral bioavailability=58% [1] The elimination half-life (t1/2) of AZD5438 is 7.2 hours in mice and 8.5 hours in rats; it is mainly metabolized in the liver, with fecal excretion accounting for 72% of the total excretion and urinary excretion accounting for 18% [1] AZD5438 is widely distributed in mice, with the drug concentration in tumor tissues being 1.8-fold of plasma concentration, and the concentrations in liver and kidney tissues being 4.3-fold and 2.9-fold of plasma concentration, respectively [1] |
| Toxicity/Toxicokinetics | The oral median lethal dose (LD50) of AZD5438 is 620 mg/kg in mice and 580 mg/kg in rats, showing low acute toxicity [1] When AZD5438 was administered orally at 100 mg/kg (once daily for 28 days), rats showed no obvious hepatotoxicity or nephrotoxicity, and the serum levels of ALT, AST, BUN, and Cr were not statistically different from those of the control group; there was no significant decrease in peripheral blood leukocyte count (change ≤±10%) [1] The human plasma protein binding rate of AZD5438 is 94%±2% [1] |
| References |
[1]. AZD5438, a potent oral inhibitor of cyclin-dependent kinases 1, 2, and 9, leads to pharmacodynamic changes and potent antitumor effects in human tumor xenografts. Mol Cancer Ther. 2009 Jul;8(7):1856-66. [2]. AZD5438, an Inhibitor of Cdk1, 2, and 9, Enhances the Radiosensitivity of Non-Small Cell Lung Carcinoma Cells. Int J Radiat Oncol Biol Phys. 2012 Nov 15;84(4):e507-14. |
| Additional Infomation |
4-(2-methyl-3-propan-2-yl-4-imidazolyl)-N-(4-methylsulfonylphenyl)-2-pyrimidinamine is a sulfonamide. AZD5438 is a potent, orally active inhibitor of CDK1/2/9 that exerts antitumor effects by inhibiting key cell cycle kinases and transcriptional regulatory kinases, inducing tumor cell cycle arrest and apoptosis [1] AZD5438 can enhance the radiosensitivity of tumor cells by inhibiting DNA damage repair pathways (such as homologous recombination repair), providing a combination therapy strategy for radiotherapy-resistant tumors [2] Preclinical studies have shown that AZD5438 is effective against various solid tumors, especially tumor types with high CDK1/2/9 activity, and is currently in the preclinical development stage [1] Purpose: Radiation therapy (RT) is one of the primary modalities for treatment of non-small cell lung cancer (NSCLC). However, due to the intrinsic radiation resistance of these tumors, many patients experience RT failure, which leads to considerable tumor progression including regional lymph node and distant metastasis. This preclinical study evaluated the efficacy of a new-generation cyclin-dependent kinase (Cdk) inhibitor, AZD5438, as a radiosensitizer in several NSCLC models that are specifically resistant to conventional fractionated RT. Methods and materials: The combined effect of ionizing radiation and AZD5438, a highly specific inhibitor of Cdk1, 2, and 9, was determined in vitro by surviving fraction, cell cycle distribution, apoptosis, DNA double-strand break (DSB) repair, and homologous recombination (HR) assays in 3 NSCLC cell lines (A549, H1299, and H460). For in vivo studies, human xenograft animal models in athymic nude mice were used. Results: Treatment of NSCLC cells with AZD5438 significantly augmented cellular radiosensitivity (dose enhancement ratio rangeing from 1.4 to 1.75). The degree of radiosensitization by AZD5438 was greater in radioresistant cell lines (A549 and H1299). Radiosensitivity was enhanced specifically through inhibition of Cdk1, prolonged G(2)-M arrest, inhibition of HR, delayed DNA DSB repair, and increased apoptosis. Combined treatment with AZD5438 and irradiation also enhanced tumor growth delay, with an enhancement factor ranging from 1.2-1.7. Conclusions: This study supports the evaluation of newer generation Cdk inhibitors, such as AZD5438, as potent radiosensitizers in NSCLC models, especially in tumors that demonstrate variable intrinsic radiation responses.[2] 4-(2-methyl-3-propan-2-yl-4-imidazolyl)-N-(4-methylsulfonylphenyl)-2-pyrimidinamine is a sulfonamide. |
Solubility Data
| Solubility (In Vitro) | DMSO : ~100 mg/mL (~269.21 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (6.73 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (6.73 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (6.73 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6921 mL | 13.4604 mL | 26.9208 mL | |
| 5 mM | 0.5384 mL | 2.6921 mL | 5.3842 mL | |
| 10 mM | 0.2692 mL | 1.3460 mL | 2.6921 mL |