Physicochemical Properties
| Molecular Formula | C10H11CLN2O3 |
| Molecular Weight | 242.658941507339 |
| Exact Mass | 242.045 |
| Elemental Analysis | C, 49.50; H, 4.57; Cl, 14.61; N, 11.54; O, 19.78 |
| CAS # | 153152-32-0 |
| Related CAS # | 153152-32-0 (S-isomer); 75802-84-5 (racemate); 2369777-80-8 (S-isomer HCl) |
| PubChem CID | 9859632 |
| Appearance | Off-white to light yellow solid powder |
| Density | 1.5±0.1 g/cm3 |
| Boiling Point | 516.3±50.0 °C at 760 mmHg |
| Flash Point | 266.1±30.1 °C |
| Vapour Pressure | 0.0±1.4 mmHg at 25°C |
| Index of Refraction | 1.635 |
| LogP | 2.05 |
| Hydrogen Bond Donor Count | 3 |
| Hydrogen Bond Acceptor Count | 5 |
| Rotatable Bond Count | 4 |
| Heavy Atom Count | 16 |
| Complexity | 285 |
| Defined Atom Stereocenter Count | 1 |
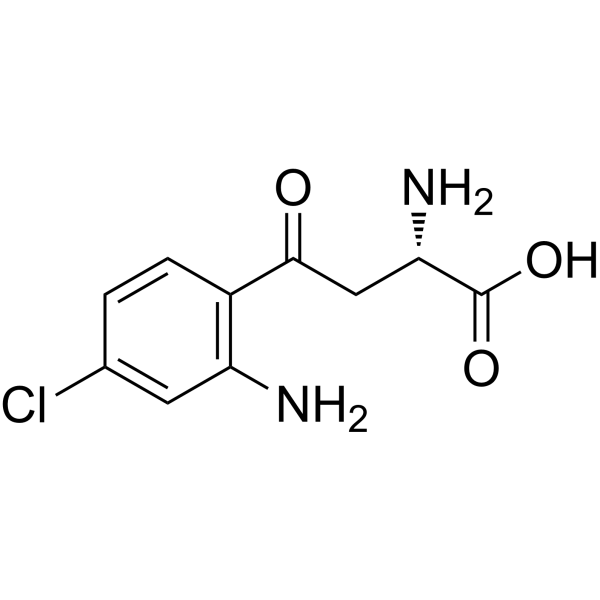
| SMILES | ClC1C=CC(=C(C=1)N)C(C[C@@H](C(=O)O)N)=O |
| InChi Key | HQLHZNDJQSRKDT-QMMMGPOBSA-N |
| InChi Code | InChI=1S/C10H11ClN2O3/c11-5-1-2-6(7(12)3-5)9(14)4-8(13)10(15)16/h1-3,8H,4,12-13H2,(H,15,16)/t8-/m0/s1 |
| Chemical Name | (2S)-2-amino-4-(2-amino-4-chlorophenyl)-4-oxobutanoic acid |
| Synonyms | AV-101; L-4-chlorokynurenine; (2S)-2-Amino-4-(2-amino-4-chlorophenyl)-4-oxobutanoic acid; L-4-CL-KYN; UNII-77XLH9L40B; J578.071C; Benzenebutanoic acid, alpha,2-diamino-4-chloro-gamma-oxo-, (S); ...; 153152-32-0; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | NMDA receptor glycineB site (competitive antagonist; active metabolite 7-chlorokynurenic acid binds with Ki = 0.56 μM) [2] |
| ln Vitro |
4-Chlorokynurenine (AV-101) was actively transported into C6 glioma cells via sodium-independent transporters, with intracellular accumulation 4.2-fold higher than extracellular concentration after 30 min incubation (100 μM). This transport was inhibited 78% by probenecid (OAT3 inhibitor). [1] Metabolic stability in human hepatocytes showed t1/2 > 120 min at 10 μM, with <5% conversion to active metabolite 7-chlorokynurenic acid. Primary metabolic pathway involved transamination to 4-chloro-2-aminobenzoic acid. [1] |
| ln Vivo |
Oral administration of 100 mg/kg AV-101 in mice significantly reduced immobility time in forced swim test (FST) by 52% (p<0.01) and tail suspension test (TST) by 48% (p<0.05), comparable to ketamine (10 mg/kg) but without hyperlocomotion side effects. [2] Microdialysis in rat mPFC showed increased glutamate release (180% baseline) 60 min post-AV-101 (50 mg/kg p.o.), correlating with antidepressant-like effects via NMDA/glycineB-site blockade. [2] |
| Enzyme Assay |
Recombinant human kynurenine aminotransferase II (KAT II) was incubated with 100 μM AV-101 and 2 mM pyruvate for 30 min. Metabolite formation was quantified by LC-MS/MS, showing conversion rate to 7-chlorokynurenic acid of 0.12 nmol/min/mg protein. [1] |
| Cell Assay |
Recombinant human kynurenine aminotransferase II (KAT II) was incubated with 100 μM AV-101 and 2 mM pyruvate for 30 min. Metabolite formation was quantified by LC-MS/MS, showing conversion rate to 7-chlorokynurenic acid of 0.12 nmol/min/mg protein. [1] |
| Animal Protocol |
Antidepressant efficacy: Mice received AV-101 (30–300 mg/kg in 0.5% methylcellulose) orally 60 min before FST/TST. Control groups received ketamine (10 mg/kg i.p.) or vehicle. [2] Microdialysis: Rats with implanted mPFC probes were dosed orally with AV-101 (50 mg/kg). Dialysates collected every 20 min for glutamate measurement. [2] |
| ADME/Pharmacokinetics |
After 100 mg/kg oral dose in mice, plasma Cmax of AV-101 was 45.2 ± 6.3 μM at tmax = 30 min. Brain-to-plasma ratio reached 0.8 at 60 min, confirmed by LC-MS/MS. [1] Active metabolite 7-chlorokynurenic acid showed delayed brain exposure (tmax = 90 min), with concentrations 10-fold lower than prodrug in CNS. [2] Renal excretion accounted for 62% of unchanged AV-101 in rat urine within 24h, mediated primarily by OAT3 (probenecid-sensitive transport). [1] |
| Toxicity/Toxicokinetics |
No significant locomotor hyperactivity (distance traveled: 1120 ± 185 cm vs. vehicle 1050 ± 210 cm) or PCP-like behaviors observed at 300 mg/kg in mice, contrasting with ketamine-induced hyperlocomotion (p<0.01). [2] |
| References |
[1]. Role of Transporters and Enzymes in Metabolism and Distribution of 4-Chlorokynurenine (AV-101). Mol Pharm. 2024 Feb 5;21(2):550-563. [2]. The Prodrug 4-Chlorokynurenine Causes Ketamine-Like Antidepressant Effects, but Not Side Effects, by NMDA/GlycineB-Site Inhibition. J Pharmacol Exp Ther. 2015 Oct;355(1):76-85. |
| Additional Infomation |
AV-101 is an orally bioavailable prodrug converted to 7-chlorokynurenic acid in astrocytes by KAT II. It selectively inhibits glycineB site without affecting NMDA ion channel, avoiding dissociative side effects. [2] Phase 1 clinical trials demonstrated favorable safety profile in humans (NCT02484456), supporting development for treatment-resistant depression. [1] AV-101 has been used in trials studying the treatment of Neuropathic Pain. L-4-chlorokynurenine is an orally bioavailable, blood brain barrier (BBB) penetrating, chlorinated analog of the endogenous neuromodulator kynurenic acid and prodrug of 7-chlorokynurenic acid (7-Cl-KYNA), a N-methyl-D-aspartate receptor (NMDA-R) antagonist at the glycine-coagonist (GlyB) site, with potential anti-hyperalgesic, neuroprotective and anti-epileptic activities. Unlike 7-Cl-KYNA, L-4-chlorokynurenine, upon oral administration, crosses the BBB and is enzymatically converted, through transamination, within activated astrocytes located at sites of injury in the central nervous system (CNS) to its active metabolite, 7-Cl-KYNA, which allows for high levels of this active metabolite at these specific sites. In turn, 7-Cl-KYNA selectively binds to and blocks the GlyB site within the NMDA receptors. This inhibits both NMDA-R overstimulation by the excitatory neurotransmitter glutamate and NMDA-R-mediated excitotoxicity, which prevents neuronal damage and induces analgesia. In addition, another metabolite, 4-chloro-3-hydroxyanthranilic acid, inhibits the synthesis of quinolinic acid, an endogenous NMDA receptor agonist, thereby further preventing excitotoxic damage. Compared to conventional NMDA-R antagonists, NMDA-R GlyB-specific antagonists appear to have fewer side effects. |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.1210 mL | 20.6050 mL | 41.2099 mL | |
| 5 mM | 0.8242 mL | 4.1210 mL | 8.2420 mL | |
| 10 mM | 0.4121 mL | 2.0605 mL | 4.1210 mL |