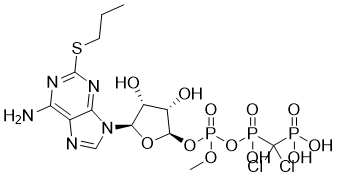
AR-C67085 is a novel and potent ADP receptor antagonist
Physicochemical Properties
| Molecular Formula | C14H22CL2N5O12P3S |
| Molecular Weight | 648.243062496185 |
| Exact Mass | 646.957 |
| Elemental Analysis | C, 25.94; H, 3.42; Cl, 10.94; N, 10.80; O, 29.62; P, 14.33; S, 4.95 |
| CAS # | 164992-25-0 |
| PubChem CID | 5310954 |
| Appearance | Typically exists as solid at room temperature |
| LogP | -2.9 |
| Hydrogen Bond Donor Count | 7 |
| Hydrogen Bond Acceptor Count | 17 |
| Rotatable Bond Count | 11 |
| Heavy Atom Count | 37 |
| Complexity | 969 |
| Defined Atom Stereocenter Count | 4 |
| SMILES | CCCSC1=NC(=C2C(=N1)N(C=N2)[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(O)OP(=O)(C(P(=O)(O)O)(Cl)Cl)O)O)O)N |
| InChi Key | ZLIAJZQKKBOFJR-WOUKDFQISA-N |
| InChi Code | InChI=1S/C14H22Cl2N5O12P3S/c1-2-3-37-13-19-10(17)7-11(20-13)21(5-18-7)12-9(23)8(22)6(32-12)4-31-36(29,30)33-35(27,28)14(15,16)34(24,25)26/h5-6,8-9,12,22-23H,2-4H2,1H3,(H,27,28)(H,29,30)(H2,17,19,20)(H2,24,25,26)/t6-,8-,9-,12-/m1/s1 |
| Chemical Name | [[[[(2R,3S,4R,5R)-5-(6-amino-2-propylsulfanylpurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl]-dichloromethyl]phosphonic acid |
| Synonyms | AR C67085; ARC67085; AR-C67085; AR-C67085; 164992-25-0; [[[[(2R,3S,4R,5R)-5-(6-amino-2-propylsulfanylpurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl]-dichloromethyl]phosphonic acid; CHEMBL336292; AR-C67085MX; PSB-0413; (((((((2R,3S,4R,5R)-5-(6-Amino-2-(propylthio)-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(hydroxy)phosphoryl)oxy)(hydroxy)phosphoryl)dichloromethyl)phosphonic acid; PSB 0413; PSB0413; PSB-0413; FPL 67085; FPL 67085XX |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | P2T receptor (pIC50 = 8.60) |
| ln Vitro | The platelet P2T receptor plays a major role in platelet aggregation, and its antagonists are predicted to have significant therapeutic potential as antithrombotic agents. We have explored analogues of adenosine triphosphate (ATP), which is a weak, nonselective but competitive P2T receptor antagonist. Modification of the polyphosphate side chain to prevent breakdown to the agonist adenosine diphosphate (ADP) and substitution of the adenine moiety to enhance affinity and selectivity for the P2T receptor led to the identification of 10e (AR-C67085 (PSB-0413; AR-C67085MX; FPL-67085) ), having an IC50 of 2.5 nM against ADP-induced aggregation of human platelets. Compound 10e was the first very potent antagonist of the P2T receptor, with a selectivity for that subtype of the P2 receptor family of >1000-fold. Further modification of the structure produced compound 10l (AR-C69931MX) having an IC50 of 0.4 nM. In vivo, at maximally effective antithrombotic doses, there is little prolongation of bleeding time (1.4-fold), which is in marked contrast to the 5-6-fold found with GPIIb/IIIa antagonists. [2] |
| ln Vivo |
In vivo studies showed that AR-C67085 (PSB-0413; AR-C67085MX; FPL-67085) had a very short duration of action, in the anesthetized rat, dog, and human (t1/2 in human approximately 2 min). In a dog model of arterial thrombosis, a full antithrombotic effect was achieved at a dose (0.1 nmol·kg-1·min-1 iv) which produced complete inhibition of platelet aggregation, measured ex vivo, and no detectable hemodynamic effects. 21 Full inhibition of aggregation was established within 15 min of commencement of infusion, and normal baseline aggregation was fully restored within 15 min of cessation of dosing. These onset/offset parameters were maintained even following continuous infusion of the compound for 6 h or more. This highly selective, rapid onset/rapid offset profile was felt to be highly desirable in the acute care setting. [2] While dialkylation of N6 of the adenine reduced receptor affinity (the N6,N6-diethyl derivative of AR-C67085 (PSB-0413; AR-C67085MX; FPL-67085) was some 1000-fold less potent than AR-C67085 (PSB-0413; AR-C67085MX; FPL-67085) ), monoalkylation gave a useful increase. A lipophilic substituent was essential (polar atoms in or upon the chain reduced receptor binding markedly: see, for example, compound 10g in Table 1), and activity increased with chain length. However, there was an effective upper limit to the length that could be tolerated, as chains of approximately four carbon atoms and longer caused the compound's biological half-life to increase sharply, unacceptably compromising the rapid onset/offset profile shown by AR-C67085 (PSB-0413; AR-C67085MX; FPL-67085) in the dog. Differences in the functional kinetics of compounds were subsequently explored in an anesthetized rat model, 22 in which ADP-induced ex vivo aggregation of blood samples taken at various time points after iv administration of compound was used to follow recovery of aggregatory behavior and hence clearance of compound. In this screen AR-C67085 (PSB-0413; AR-C67085MX; FPL-67085) showed >93% recovery 20 min after dosing. Long substituent chains on N6 reduced this to <10%, as, surprisingly, did a 2,2,2-trifluoroethyl group (see 10f,j). A methylthioethyl substituent at this position (10h) was found to enhance affinity with somewhat less effect upon half-life. In combination with a 3,3,3-trifluoropropylthio group at C-2, the N6-methylthioethyl group gave a compound with a half-life similar to that of AR-C67085 (PSB-0413; AR-C67085MX; FPL-67085) (showing 79% recovery of aggregation behavior at the 20 min time point). This compound 10l, being some 6-fold more potent than AR-C67085 (PSB-0413; AR-C67085MX; FPL-67085) , has been taken forward into development and is currently in phase II of development. [2] While profound inhibition of platelet function provides the potential for beneficial antithrombotic effects, this may be achieved at the expense of compromised hemostasis, with a subsequent increase in the risk of bleeding, particularly during invasive procedures. To assess the potential for undesirable antihemostatic effects, we have examined the relationship between the antithrombotic activity of 10l and its effect upon bleeding time in the dog. Dose−response relationships show a favorable (98-fold) separation between the desired antithrombotic action and the prolongation of bleeding time. In consequence, the full inhibition of platelet aggregation needed to give an antithrombotic effect is achieved at doses which extend bleeding time by less than 2-fold (see Figure 1a). This substantial separation of the two effects is in marked contrast to the pharmacological behavior of members of the GPIIb/IIIa antagonist class (agents currently attracting considerable interest for antithrombotic therapy). We have examined three examples (of diverse structure and profile), and each has shown a separation of a mere 2−5-fold. Figure 1b shows the data generated with the compound Lamifiban (Ro 449883), which is quite representative of our observations. Full inhibition of aggregation by these GPIIb/IIIa antagonists is achieved at dose levels such that bleeding time is prolonged by some 6−7-fold. Clearly the risk of persistent hemorrhage following administration of a P2T receptor antagonist is likely to be very much less than would be the case with such GPIIb/IIIa antagonists. We believe this substantial separation of the two effects constitutes a major advantage for the P2T antagonist mechanism of action in the modulation of platelet aggregation. [2] |
| Enzyme Assay |
Biological Assays. [2] The affinity of the test compounds for the P2T receptor was assayed using washed human platelets by the method of Humphries et al. Antithrombotic activity was measured using a cyclic flow reduction model in dog femoral artery. Functional kinetics of the compounds were determined in the anesthetized rat by the method of Humphries et al. Br. J. Pharmacol. 1994, 114, 63P. |
| References |
[1]. A novel series of P2T purinoceptor antagonists: definition of the role of ADP in arterial thrombosis. Trends Pharmacol Sci. 1995 Jun;16(6):179-81. [2]. Antagonists of the platelet P2T receptor: a novel approach to antithrombotic therapy. J Med Chem. 1999 Jan 28;42(2):213-20. |
| Additional Infomation | Modification of ATP has led to the identification of 10d (AR-C66096MX), an important pharmacological tool for the characterization of P2T receptors, and to the discovery of the therapeutically useful analogues 10e (AR-C67085 (PSB-0413; AR-C67085MX; FPL-67085) ) and 10l (AR-C69931MX). In a healthy human volunteer study with 10e, a dose-related inhibition of ex vivo ADP-induced platelet aggregation has been demonstrated, with rapid recovery on termination of the infusion and with little or no prolongation of bleeding time. The same profile has been demonstrated for 10l, which is in phase II development for the treatment of acute coronary syndromes such as unstable angina and in the setting of percutaneous transluminal coronary artery revascularization. A significant feature of the pharmacological profile of P2T receptor antagonists is the unprecedentedly advantageous separation of antithrombotic activity from effects on bleeding time. On the basis of this clear and consistent advantage, we believe antagonists of the P2T receptor represent a major step forward in the treatment of thrombotic disease. [2] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.5426 mL | 7.7132 mL | 15.4264 mL | |
| 5 mM | 0.3085 mL | 1.5426 mL | 3.0853 mL | |
| 10 mM | 0.1543 mL | 0.7713 mL | 1.5426 mL |