Abaloparatide (Trade name Tymlos; formerly known as BA-058; ITM-058; BIM 44058) is a 34 amino acid synthetic analog of parathyroid hormone-related protein (PTHrP) that is used to treat osteoporosis. The Food and Drug Administration (FDA) approved it on April 28, 2017, for the treatment of postmenopausal osteoporosis. Not unlike bisphosphonates, but similar to the related medication teriparatide, it is an anabolic (bone-growing) agent. The drug's subcutaneous injection formulation has finished a Phase III osteoporosis trial. Only this one study revealed a decline in fractures. Parathyroid hormone (PTH) (1-34) and parathyroid hormone-related protein (PTHrP) (1-34) share 76% and 41% homology, respectively, with abaloparatide. By selectively activating the parathyroid hormone 1 receptor (PTH1R), a G protein-coupled receptor (GPCR) expressed in osteoblasts and osteocytes, it functions as an anabolic agent for bone. Abaloparatide preferentially binds the PTH1R's RG conformational state, which causes a brief downstream cyclic AMP signaling response that points in the direction of a more anabolic signaling pathway.
Physicochemical Properties
| Molecular Formula | C174H300N56O49 |
| Molecular Weight | 3960.58963775635 |
| Exact Mass | 3959.273 |
| Elemental Analysis | C, 52.58; H, 7.62; N, 19.51; O, 20.29 |
| CAS # | 247062-33-5 |
| Related CAS # | Abaloparatide TFA |
| PubChem CID | 145705876 |
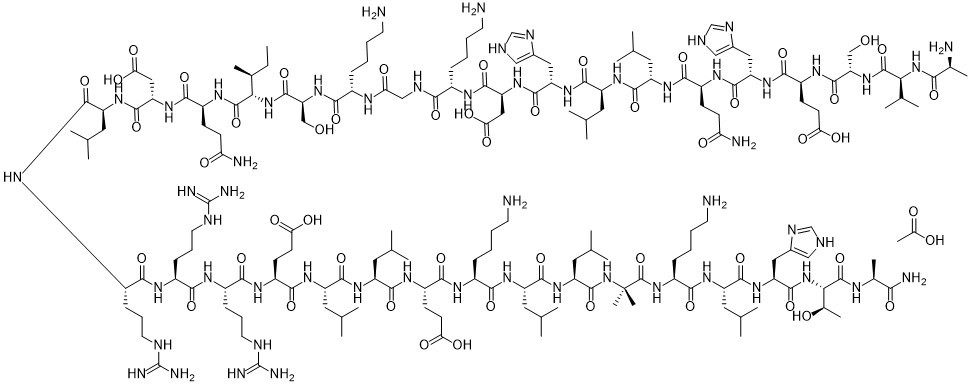
| Sequence | H-DL-Ala-DL-Val-DL-Ser-DL-Glu-DL-His-DL-Gln-DL-Leu-DL-Leu-DL-His-DL-Asp-DL-Lys-Gly-DL-Lys-DL-Ser-DL-xiIle-DL-Gln-DL-Asp-DL-Leu-DL-Arg-DL-Arg-DL-Arg-DL-Glu-DL-Leu-DL-Leu-DL-Glu-DL-Lys-DL-Leu-DL-Leu-Aib-DL-Lys-DL-Leu-DL-His-DL-xiThr-DL-Ala-NH2 |
| SequenceShortening | AVSEHQLLHDKGKSXQDLRRRELLEKLLXKLHXA; AVSEHQLLHDKGKSIQDLRRRELLEKLL-{Aib}-KLHTA-NH2 |
| Appearance | White to off-white solid powder |
| LogP | -20.9 |
| Hydrogen Bond Donor Count | 61 |
| Hydrogen Bond Acceptor Count | 60 |
| Rotatable Bond Count | 145 |
| Heavy Atom Count | 279 |
| Complexity | 9310 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | BVISQZFBLRSESR-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C174H300N56O49/c1-26-93(20)136(228-165(274)126(80-232)224-141(250)101(39-28-32-56-176)200-129(236)78-195-140(249)100(38-27-31-55-175)201-161(270)123(73-133(243)244)223-160(269)121(71-98-76-190-82-197-98)220-158(267)118(68-90(14)15)216-155(264)114(64-86(6)7)213-148(257)107(45-50-127(180)234)207-159(268)120(70-97-75-189-81-196-97)219-151(260)111(49-54-132(241)242)209-164(273)125(79-231)225-167(276)135(92(18)19)227-139(248)94(21)179)168(277)210-108(46-51-128(181)235)149(258)222-124(74-134(245)246)162(271)217-112(62-84(2)3)152(261)205-105(44-37-61-194-173(187)188)143(252)203-103(42-35-59-192-171(183)184)142(251)204-104(43-36-60-193-172(185)186)144(253)206-110(48-53-131(239)240)150(259)214-115(65-87(8)9)154(263)215-113(63-85(4)5)153(262)208-109(47-52-130(237)238)147(256)202-102(40-29-33-57-177)145(254)211-116(66-88(10)11)156(265)218-119(69-91(16)17)166(275)230-174(24,25)170(279)226-106(41-30-34-58-178)146(255)212-117(67-89(12)13)157(266)221-122(72-99-77-191-83-198-99)163(272)229-137(96(23)233)169(278)199-95(22)138(182)247/h75-77,81-96,100-126,135-137,231-233H,26-74,78-80,175-179H2,1-25H3,(H2,180,234)(H2,181,235)(H2,182,247)(H,189,196)(H,190,197)(H,191,198)(H,195,249)(H,199,278)(H,200,236)(H,201,270)(H,202,256)(H,203,252)(H,204,251)(H,205,261)(H,206,253)(H,207,268)(H,208,262)(H,209,273)(H,210,277)(H,211,254)(H,212,255)(H,213,257)(H,214,259)(H,215,263)(H,216,264)(H,217,271)(H,218,265)(H,219,260)(H,220,267)(H,221,266)(H,222,258)(H,223,269)(H,224,250)(H,225,276)(H,226,279)(H,227,248)(H,228,274)(H,229,272)(H,230,275)(H,237,238)(H,239,240)(H,241,242)(H,243,244)(H,245,246)(H4,183,184,192)(H4,185,186,193)(H4,187,188,194) |
| Chemical Name | Ala-Val-Ser-Glu-His-Gln-Leu-Leu-His-Asp-Lys-Gly-Lys-Ser-Ile-Gln-Asp-Leu-Arg-ArgArg-Glu-Leu-Leu-Glu-Lys-Leu-Leu-Aib-Lys-Leu-His-Thr-Ala-NH2 |
| Synonyms | BIM-44058; Abaloparatide acetate; BA-058; ITM-058; BIM 44058; BA 058;ITM 058; BIM44058; BA058;ITM058; H-DL-Ala-DL-Val-DL-Ser-DL-Glu-DL-His-DL-Gln-DL-Leu-DL-Leu-DL-His-DL-Asp-DL-Lys-Gly-DL-Lys-DL-Ser-DL-xiIle-DL-Gln-DL-Asp-DL-Leu-DL-Arg-DL-Arg-DL-Arg-DL-Glu-DL-Leu-DL-Leu-DL-Glu-DL-Lys-DL-Leu-DL-Leu-Aib-DL-Lys-DL-Leu-DL-His-DL-xiThr-DL-Ala-NH2; trade name: Tymlos |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment (e.g. under nitrogen), avoid exposure to moisture and light. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
PTHR1/parathyroid hormone receptor 1 Abaloparatide (0-100 nM; 40 min) increases β-arrestin recruitment and Gs/cAMP signaling in MC3T3-E1 cells[1]. Abaloparatide (0-100 nM) effectively and dose-dependently induces PTHR1 internalization in U2OS cells, with an EC50 value of 0.8 nM[1]. |
| ln Vitro |
Abaloparatide (0-100 nM; 40 min) increases β-arrestin recruitment and Gs/cAMP signaling in MC3T3-E1 cells[1]. Abaloparatide (0-100 nM) effectively and dose-dependently induces PTHR1 internalization in U2OS cells, with an EC50 value of 0.8 nM[1]. In MC3T3-E1 osteoblastic cells, abaloparatide stimulated intracellular cAMP production in a dose-dependent manner (0.01-100 nM), with an EC50 of 0.3 ± 0.03 nM, which was 2.3-fold lower (more potent) than that of teriparatide (EC50 = 0.7 ± 0.2 nM). The maximum cAMP stimulation was not significantly different between the two analogs. [1] In a PathHunter β-arrestin recruitment assay using CHO-K1 cells expressing PTHR1, abaloparatide induced a dose-dependent increase in β-arrestin/PTHR1 interaction, with an EC50 of 0.9 ± 0.2 nM. This EC50 was 1.6-fold lower than that of teriparatide (EC50 = 1.5 ± 0.3 nM). The maximal stimulation for both was reached at 100 nM. [1] In a PathHunter PTHR1 internalization assay using U2OS cells, both abaloparatide and teriparatide efficiently induced dose-dependent receptor internalization. The EC50 values for abaloparatide (0.8 ± 0.4 nM) and teriparatide (1.1 ± 0.4 nM) were not statistically different. [1] |
| ln Vivo |
In mice, abelaparatide (20–80 µg/kg; s.c.; daily for 30 days) improves cortical structure and bone formation[1]. Teriparatide and abaloparatide are parathyroid hormone receptor 1 (PTHR1) analogs with unexplained differential efficacy for the treatment of osteoporosis. Therefore, we compared the effects of abaloparatide and teriparatide on bone structure, turnover, and levels of receptor activator of nuclear factor-kappa B ligand (RANKL) and osteoprotegerin (OPG). Wild-type (WT) female mice were injected daily with vehicle or 20-80 µg/kg/day of teriparatide or abaloparatide for 30 days. Femurs and spines were examined by microcomputed tomography scanning and serum levels of bone turnover markers, RANKL, and OPG, were measured by ELISA. Both analogs similarly increased the distal femoral fractional trabecular bone volume, connectivity, and number, and reduced the structure model index (SMI) at 20-80 µg/kg/day doses. However, only abaloparatide exhibited a significant increase (13%) in trabecular thickness at 20 µg/kg/day dose. Femoral cortical evaluation showed that abaloparatide caused a greater dose-dependent increase in cortical thickness than teriparatide. Both teriparatide and abaloparatide increased lumbar 5 vertebral trabecular connectivity but had no or modest effect on other indices. Biochemical analysis demonstrated that abaloparatide promoted greater elevation of procollagen type 1 intact N-terminal propeptide, a bone formation marker, and tartrate-resistant acid phosphatase 5b levels, a bone resorption marker, and lowered the RANKL/OPG ratio. Furthermore, PTHR1 signaling was compared in cells treated with 0-100 nmol/L analog. Interestingly, abaloparatide had a markedly lower EC50 for cAMP formation (2.3-fold) and β-arrestin recruitment (1.6-fold) than teriparatide. Therefore, abaloparatide-improved efficacy can be attributed to enhanced bone formation and cortical structure, reduced RANKL/OPG ratio, and amplified Gs-cAMP and β-arrestin signaling.[1] Abaloparatide is a novel 34-amino acid peptide selected to be a potent and selective activator of the parathyroid hormone receptor (PTH1R) signaling pathway with 41% homology to PTH(1-34) and 76% homology to PTHrP(1-34). A 12-month treatment study was conducted in osteopenic ovariectomized (OVX) rats to characterize the mechanisms by which abaloparatide increases bone mass. Sprague-Dawley (SD) rats were subjected to OVX or sham surgery at age 6 months and left untreated for 3 months to allow OVX-induced bone loss. Ten OVX rats were euthanized after this bone depletion period, and the remaining OVX rats received daily subcutaneous injections of vehicle (n = 18) or abaloparatide at 1, 5, or 25 μg/kg/d (n = 18/dose level) for 12 months. Sham controls (n = 18) received vehicle daily. Bone densitometry and biochemical markers of bone formation and resorption were assessed longitudinally, and L3 vertebra and tibia were collected at necropsy for histomorphometry. Abaloparatide increased biochemical bone formation markers without increasing bone resorption markers or causing hypercalcemia. Abaloparatide increased histomorphometric indices of bone formation on trabecular, endocortical, and periosteal surfaces without increasing osteoclasts or eroded surfaces. Abaloparatide induced substantial increases in trabecular bone volume and density and improvements in trabecular microarchitecture. Abaloparatide stimulated periosteal expansion and endocortical bone apposition at the tibial diaphysis, leading to marked increases in cortical bone volume and density. Whole-body bone mineral density (BMD) remained stable in OVX-Vehicle controls while increasing 25% after 12 months of abaloparatide (25 μg/kg). Histomorphometry and biomarker data suggest that gains in cortical and trabecular bone mass were attributable to selective anabolic effects of abaloparatide, without evidence for stimulated bone resorption. © 2016 American Society for Bone and Mineral Research.[2] In 16-week-old female C57BL/6J mice treated with daily subcutaneous injections of abaloparatide (20 or 80 µg/kg/day) for 30 days, femoral trabecular bone thickness (Tb.Th) was significantly increased (113% and 108% of vehicle, respectively) compared to vehicle controls, while teriparatide did not significantly increase Tb.Th at 20 µg/kg/day. Both peptides similarly improved other femoral trabecular parameters (BV/TV, Conn.D, SMI). [1] In the same mouse model, abaloparatide at both 20 and 80 µg/kg/day significantly increased cortical thickness (Ct.Th) at the femoral midshaft by 17% and 18%, respectively. Teriparatide significantly increased Ct.Th only at the 80 µg/kg/day dose. [1] Serum analysis showed that abaloparatide treatment produced a significant, dose-dependent increase in the bone formation marker P1NP (227% and 407% of vehicle at 20 and 80 µg/kg/day, respectively). Teriparatide significantly increased P1NP only at 80 µg/kg/day (291%). [1] Abaloparatide treatment increased serum levels of the osteoclast activity marker TRAcP-5b at both 20 (167%) and 80 µg/kg/day (227%), while teriparatide increased it only at 80 µg/kg/day (193%). Neither peptide altered serum levels of the resorption marker CTX. [1] Abaloparatide at 80 µg/kg/day increased serum osteoprotegerin (OPG) levels by 44% and significantly decreased the RANKL/OPG ratio by 42% compared to vehicle. Teriparatide did not significantly alter OPG or the RANKL/OPG ratio. [1] No significant differences in serum calcium (Ca²⁺) levels were found among vehicle, abaloparatide, and teriparatide groups when compared to each other. However, abaloparatide groups (20 & 80 µg/kg) showed a 24% increase in Ca²⁺ compared to vehicle alone. Both peptides at 80 µg/kg/day increased serum inorganic phosphate (Pi) levels by 50%. [1] |
| Enzyme Assay |
PathHunter® eXpress PTHR1 CHO‐K1 β‐arrestin GPCR assay[1] To assess the effects of Abaloparatide and teriparatide stimulation of PTHR1 on β‐arrestin recruitment to the cell membrane, a PathHunter eXpress PTHR1 Chinese Hamster Ovary‐K1 (CHO‐K1) β‐arrestin GPCR Assay was used. The assay takes advantage of Enzyme Fragment Complementation technology. The PTHR1 is fused in frame with a small enzyme donor fragment ProLink™ (PK) and co‐expressed in CHO‐K1 cells stably expressing a fusion protein of β‐arrestin and the larger, N‐terminal deletion mutant of β‐galactosidase (called enzyme acceptor or EA). Activation of the PTHR1 stimulates binding of β‐arrestin to the PK‐tagged GPCR and forces complementation of the two enzyme fragments, resulting in the formation of an active β‐galactosidase enzyme. An increase in enzyme activity is then measured using chemiluminescent PathHunter Detection Reagents. Cell seeding, incubation, and detection were performed as instructed by the manufacturer. Briefly, cells were seeded in a clear bottom white 96‐well plate and incubated for 48 h at 37°C CO2 incubator. Cells were treated with vehicle, teriparatide, or Abaloparatide for 60 min at 37°C in a CO2 incubator. At the end of the incubation, β‐gal enzyme substrate was added for 60 min at room temperature in the dark. Light generation (Relative Light Units, RLU), an indication of β‐gal enzyme fragment complementation and β‐Arrestin/ PTHR1 interaction, was measured using BMG Labtech PHERAstar FS luminescence plate reader.[1] PathHunter® eXpress PTHR1 activated GPCR internalization assay[1] To determine PTHR1 internalization, we used PathHunter eXpress PTHR1 U2OS Activated GPCR Internalization Assay. PathHunter® PTHR1 Activated GPCR Internalization U2OS cell lines are engineered to co‐express an untagged PTHR1, an EA‐tagged β‐arrestin, and a PK tag localized to the endosomes. Activation of the untagged PTHR1 induces β‐arrestin recruitment, followed by internalization of the GPCR‐β‐arrestin‐EA complex in PK‐tagged endosomes. Similar to the β‐arrestin assay format, this internalization forces complementation of the two β‐gal enzyme fragments, forming functional enzyme that hydrolyzes substrate to generate a chemiluminescent signal. U2OS osteoblastic cell line seeding, incubation, and detection were performed as instructed by the manufacturer. Cells were treated with vehicle, teriparatide, or Abaloparatide for 60 min at 37°C in a CO2 incubator. At the end of the incubation, β‐gal enzyme substrate was added for 60 min at room temperature in the dark. Light generation (RLU), an indication of β‐gal enzyme fragment complementation and β‐arrestin/endosome/PTHR1 formation, was measured using BMG Labtech PHERAstar FS luminescence plate reader. |
| Cell Assay |
Measurement of intracellular cAMP generation[1] MC3T3‐E1 cells were seeded at 40,000 cells/well of a 24‐well plate containing 500‐µL alpha‐MEM supplemented with 10% FBS and 1% PS. After culture for 1 week, the medium was removed and replaced with 250 µL of stimulation medium (alpha‐MEM containing 0.05% FBS, 0.1% BSA, 5 mmol/L hepes buffer, and 0.5 mmol/L IBMX) for 15 min. IBMX is a phosphodiesterase inhibitor that prevents degradation of the generated cAMP. Vehicle, Abaloparatide, and teriparatide were then added in 250 µL stimulation medium to achieve final concentrations of 0, 0.01, 0.1, 1, 10, and 100 nmol/L/well. Incubation continued for 40 min at 37°C before the medium was removed and the plates were snap frozen in liquid N3 and stored at −80°C. For extraction of intracellular cAMP, 100 mmol/L Hcl was added and cells were incubated at room temperature for 1 h. Intracellular cAMP was assayed using a cAMP competitive ELISA kit and following the manufacturer protocol and instructions. For intracellular cAMP measurement, MC3T3-E1 cells were seeded in 24-well plates and cultured for one week. The medium was replaced with a stimulation medium containing a phosphodiesterase inhibitor. Cells were then treated with vehicle or various concentrations of abaloparatide or teriparatide (0-100 nM) for 40 minutes at 37°C. The reaction was stopped by snap-freezing, and intracellular cAMP was extracted with HCl and quantified using a competitive ELISA. [1] For β-arrestin recruitment assessment, a commercial cell-based assay utilizing enzyme fragment complementation technology was employed. Cells expressing a PTHR1-prolink tag fusion and β-arrestin-enzyme acceptor fusion were seeded in 96-well plates. After 48 hours, cells were treated with vehicle or peptides (0-100 nM) for 60 minutes at 37°C. A chemiluminescent substrate was added, and the signal (Relative Light Units, RLU), indicative of β-arrestin/PTHR1 interaction, was measured. [1] For PTHR1 internalization assessment, a similar commercial cell-based assay was used with a cell line engineered to co-express untagged PTHR1, β-arrestin-enzyme acceptor, and a prolink tag localized to endosomes. Cells were treated with vehicle or peptides (0-100 nM) for 60 minutes at 37°C. Internalization of the receptor complex into labeled endosomes forces enzyme fragment complementation, which was detected by adding a chemiluminescent substrate and measuring RLU. [1] |
| Animal Protocol |
16-week-old wild-type (WT) female C57BL/6J mice[1] 20-80 µg/kg S.c.; daily for 30 days All experiments were conducted on 16‐week‐old wild‐type (WT) female C57BL/6J mice (Stock number 664). Vehicle (0.9% NaCl/10 mmol/L acetic acid) or 20–80 µg/kg/day teriparatide or abaloparatide was injected subcutaneously (SC) daily (except Sunday) and continued for 30 days. No peptide injection was performed on the day of animal sacrifice.[1] A total of 13 rats were euthanized or found dead between study days 117 to 358 before study termination: 6 in the sham control group, 2 in the OVX-Veh group, 3 in the OVX + abaloparatide 1 μg/kg/d group, 1 in the OVX + abaloparatide 5 μg/kg/d group, and 1 in the OVX + abaloparatide 25 μg/kg/d group. For these animals’ data, absolute values were reported if collected, and data based on % change from baseline were censored as required. Five deaths were likely secondary to complications from blood collection, whereas the remaining deaths were attributed to incidental age-related pathologies.[1] Study design and dose selection[1] After a 13-week postsurgical bone depletion period, one group of untreated OVX rats was euthanized as a pretreatment baseline group for histomorphometry data. The remaining groups were given daily s.c. injections of vehicle (Vehicle; 0.9% sodium chloride) or one of three dose levels of abaloparatide in a 0.1 mL/kg volume. Abaloparatide dose levels were 1 μg/kg/d (OVX-ABL1), 5 μg/kg/d (OVX-ABL5), and 25 μg/kg/d (OVX-ABL25), with dosing guided by weekly body weight measurements. Preliminary results from another rat study indicated that 6 weeks of abaloparatide at 1.25 μg/kg/d completely reversed OVX-induced bone loss (Radius Health, Inc., Waltham, MA, USA). This led to selection of 1 μg/kg as the low dose, and also 5- and 25-fold multiples of this dose to provide safety margins.[1] Sixteen-week-old female wild-type C57BL/6J mice were used. Animals were randomized into groups receiving daily subcutaneous injections (except Sundays) for 30 days of: vehicle (0.9% NaCl/10 mmol/L acetic acid), abaloparatide (20 or 80 µg/kg/day), or teriparatide (20 or 80 µg/kg/day). No injection was given on the day of sacrifice. [1] At the end of the treatment period, mice were anesthetized, and blood was collected via cardiac puncture for serum isolation. Animals were then euthanized, and femurs and lumbar vertebrae (L5) were harvested, fixed in formalin, and preserved in ethanol for micro-computed tomography (micro-CT) analysis to assess bone microarchitecture. [1] |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion The absolute bioavailability of abaloparatide in healthy women after subcutaneous administration of an 80 mcg dose was 36%. Following subcutaneous administration of 80 mcg abaloparatide in postmenopausal women with osteoporosis for seven days, the mean (SD) Cmax was 812 (118) pg/mL and the AUC0-24 was 1622 (641) pgxhr/mL. The median Tmax was 0.51 hours, with a range from 0.25 to 0.52 hours. The peptide fragments of abaloparatide are primarily eliminated through renal excretion. The volume of distribution was approximately 50 L. The mean apparent total plasma clearance for subcutaneous administration is 168 L/h in healthy subjects. Metabolism / Metabolites Abaloparatide is metabolized into smaller peptide fragments via non-specific proteolytic degradation. Biological Half-Life The mean half-life of abaloparatide is approximately one hour. |
| Toxicity/Toxicokinetics |
Protein Binding _In vitro_, abaloparatide was approximately 70% bound to plasma proteins. Daily subcutaneous administration of abaloparatide at doses up to 25 µg/kg/d for 12 months was well-tolerated in OVX rats, with no observed weight loss or signs of toxicity compared to vehicle controls. Abaloparatide did not cause meaningful hypercalcemia; serum calcium levels remained within the normal range throughout the study, with only a marginal (+2.6%) increase at month 6 for the 25 µg/kg/d group. The highest recorded serum calcium value was 11.7 mg/dL (within normal range). Uterine and vaginal weights were not influenced by abaloparatide treatment. [2] |
| References |
[1]. Abaloparatide exhibits greater osteoanabolic response and higher cAMP stimulation and β-arrestin recruitment than teriparatide. Physiol Rep. 2019 Oct;7(19):e14225. [2]. One Year of Abaloparatide, a Selective Activator of the PTH1 Receptor, Increased Bone Formation and Bone Mass in Osteopenic Ovariectomized Rats Without Increasing Bone Resorption. J Bone Miner Res. 2017 Jan;32(1):24-33. |
| Additional Infomation |
Pharmacodynamics Abaloparatide stimulates bone formation on periosteal, trabecular, and cortical bone surfaces. It increases bone mineral density and bone formation markers in a dose-dependent manner. Abaloparatide causes transient and limited increases in osteoclast bone resorption and increases bone density. In rats and monkeys, abaloparatide exerted anabolic effects, increasing bone mineral density and mineral content correlating with increases in bone strength at vertebral and nonvertebral sites. Abaloparatide is a synthetic analog of parathyroid hormone-related protein (PTHrP 1-34) with 71% homology to PTHrP(1-34) and 41% homology to teriparatide (PTH 1-34). It contains modifications in five amino acids between residues 22 and 34. [1] Both abaloparatide and teriparatide are agonists of the PTH1 receptor (PTHR1) and are FDA-approved for the treatment of osteoporosis. The study suggests that the superior osteoanabolic effects of abaloparatide may be attributed to its more potent activation of both Gs/cAMP and β-arrestin signaling pathways downstream of PTHR1 compared to teriparatide. [1] The enhanced signaling leads to greater stimulation of bone formation (higher P1NP), improved cortical and trabecular bone thickness, and a potentially more favorable bone resorption profile (lower RANKL/OPG ratio) in the tested mouse model. [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (0.63 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (0.63 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (0.63 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 0.2525 mL | 1.2624 mL | 2.5249 mL | |
| 5 mM | 0.0505 mL | 0.2525 mL | 0.5050 mL | |
| 10 mM | 0.0252 mL | 0.1262 mL | 0.2525 mL |